Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement
Endocrine Section of First Department of Medicine (E.D.-K.), Laiko Hospital, Medical School University of Athens, 11527 Athens, Greece; Department of Pediatrics (J.-P.B.), Centre Hospitalier Universitaire de Liege, 4000 Liege, Belgium; Department of Obstetrics, Gynecology, and Reproductive Sciences (L.C.G.), University of California San Francisco, San Francisco, California 94131; Department of Environmental Health (R.H.), Harvard School of Public Health, Boston, Massachusetts 02115; Department of Urology (G.S.P.), University of Illinois at Chicago, Chicago, Illinois 60612; Department of Anatomy and Cell Biology (A.M.S.), Tufts University School of Medicine, Boston, Massachusetts 02111; Biology Department (R.T.Z.), University of Massachusetts, Amherst, Massachusetts 01003; and Division of Pharmacology and Toxicology (A.C.G.), The University of Texas at Austin, Austin, Texas 78712
Address all correspondence and requests for reprints to: Andrea C. Gore, Ph.D., The University of Texas at Austin, College of Pharmacy, 1 University Station, A1915, Austin, Texas 78712. E-mail: ude.saxetu.liam@erog.aerdna.
Abstract
There is growing interest in the possible health threat posed by endocrine-disrupting chemicals (EDCs), which are substances in our environment, food, and consumer products that interfere with hormone biosynthesis, metabolism, or action resulting in a deviation from normal homeostatic control or reproduction. In this first Scientific Statement of The Endocrine Society, we present the evidence that endocrine disruptors have effects on male and female reproduction, breast development and cancer, prostate cancer, neuroendocrinology, thyroid, metabolism and obesity, and cardiovascular endocrinology. Results from animal models, human clinical observations, and epidemiological studies converge to implicate EDCs as a significant concern to public health. The mechanisms of EDCs involve divergent pathways including (but not limited to) estrogenic, antiandrogenic, thyroid, peroxisome proliferator-activated receptor γ, retinoid, and actions through other nuclear receptors; steroidogenic enzymes; neurotransmitter receptors and systems; and many other pathways that are highly conserved in wildlife and humans, and which can be modeled in laboratory in vitro and in vivo models. Furthermore, EDCs represent a broad class of molecules such as organochlorinated pesticides and industrial chemicals, plastics and plasticizers, fuels, and many other chemicals that are present in the environment or are in widespread use. We make a number of recommendations to increase understanding of effects of EDCs, including enhancing increased basic and clinical research, invoking the precautionary principle, and advocating involvement of individual and scientific society stakeholders in communicating and implementing changes in public policy and awareness.
I. General Introduction to Endocrine Disruption
An endocrine-disrupting compound was defined by the U.S. Environmental Protection Agency (EPA) as “an exogenous agent that interferes with synthesis, secretion, transport, metabolism, binding action, or elimination of natural blood-borne hormones that are present in the body and are responsible for homeostasis, reproduction, and developmental process.” Our understanding of the mechanisms by which endocrine disruptors exert their effect has grown. Endocrine-disrupting chemicals (EDCs) were originally thought to exert actions primarily through nuclear hormone receptors, including estrogen receptors (ERs), androgen receptors (ARs), progesterone receptors, thyroid receptors (TRs), and retinoid receptors, among others. Today, basic scientific research shows that the mechanisms are much broader than originally recognized. Thus, endocrine disruptors act via nuclear receptors, nonnuclear steroid hormone receptors (e.g., membrane ERs), nonsteroid receptors (e.g., neurotransmitter receptors such as the serotonin receptor, dopamine receptor, norepinephrine receptor), orphan receptors [e.g., aryl hydrocarbon receptor (AhR)—an orphan receptor], enzymatic pathways involved in steroid biosynthesis and/or metabolism, and numerous other mechanisms that converge upon endocrine and reproductive systems. Thus, from a physiological perspective, an endocrine-disrupting substance is a compound, either natural or synthetic, which, through environmental or inappropriate developmental exposures, alters the hormonal and homeostatic systems that enable the organism to communicate with and respond to its environment.
The group of molecules identified as endocrine disruptors is highly heterogeneous and includes synthetic chemicals used as industrial solvents/lubricants and their byproducts [polychlorinated biphenyls (PCBs), polybrominated biphenyls (PBBs), dioxins], plastics [bisphenol A (BPA)], plasticizers (phthalates), pesticides [methoxychlor, chlorpyrifos, dichlorodiphenyltrichloroethane (DDT)], fungicides (vinclozolin), and pharmaceutical agents [diethylstilbestrol (DES)].
Natural chemicals found in human and animal food (e.g., phytoestrogens, including genistein and coumestrol) can also act as endocrine disruptors. These substances, whereas generally thought to have relatively low binding affinity to ERs, are widely consumed and are components of infant formula (1,2). A recent study reported that urinary concentrations of the phytoestrogens genistein and daidzein were about 500-fold higher in infants fed soy formula compared with those fed cow’s milk formula (3). Therefore, the potential for endocrine disruption by phytoestrogens needs to be considered.
The sources of exposure to EDCs are diverse and vary widely around the world. The situation is constantly evolving because some EDCs were banned decades ago and others more recently, with significant differences between countries. In this respect, migrating people provide a model to study cessation and/or onset of exposure depending on contamination of the original and new milieus. There are also several historical examples of toxic spills or contamination from PCBs and dioxins that show a direct causal relationship between a chemical and the manifestation of an endocrine or reproductive dysfunction in humans and wildlife. However, these types of single exposures are not representative of more common widespread persistent exposure to a broad mix of indoor and outdoor chemicals and contaminants. Industrialized areas are typically characterized by contamination from a wide range of industrial chemicals that may leach into soil and groundwater. These complex mixtures enter the food chain and accumulate in animals higher up the food chain such as humans, American bald eagles, polar bears, and other predatory animals. Exposure occurs through drinking contaminated water, breathing contaminated air, ingesting food, or contacting contaminated soil. People who work with pesticides, fungicides, and industrial chemicals are at particularly high risk for exposure and thus for developing a reproductive or endocrine abnormality.
Some EDCs were designed to have long half-lives; this was beneficial for their industrial use, but it has turned out to be quite detrimental to wildlife and humans. Because these substances do not decay easily, they may not be metabolized, or they may be metabolized or broken down into more toxic compounds than the parent molecule; even substances that were banned decades ago remain in high levels in the environment, and they can be detected as part of the body burden of virtually every tested individual animal or human (4,5). In fact, some endocrine disruptors are detectable in so-called “pristine” environments at remote distances from the site they were produced, used, or released due to water and air currents and via migratory animals that spend part of their life in a contaminated area, to become incorporated into the food chain in an otherwise uncontaminated region. Others, such as BPA, may not be as persistent [although recent evidence (e.g., Ref. 6) suggests longer half-lives) but are so widespread in their use that there is prevalent human exposure.
A. Important issues in endocrine disruption
A number of issues have proven to be key to a full understanding of mechanisms of action and consequences of exposure to EDCs. These have been reviewed previously in detail (7), and several of them are listed here in brief.
1. Age at exposure
Exposure of an adult to an EDC may have very different consequences from exposure to a developing fetus or infant. In fact, the field of endocrine disruption has embraced the terminology “the fetal basis of adult disease” (8) to describe observations that the environment of a developing organism, which includes the maternal environment (eutherian mammals), the egg (other vertebrates), and the external environment, interacts with the individual’s genes to determine the propensity of that individual to develop a disease or dysfunction later in life. In this Scientific Statement, we extend this concept beyond the fetal period to the early postnatal developmental period when organs continue to undergo substantial development. Thus, we will henceforward use the terminology “the developmental basis of adult disease.”
2. Latency from exposure
The developmental basis of adult disease also has implicit in its name the concept that there is a lag between the time of exposure and the manifestation of a disorder. In other words, consequences of developmental exposure may not be immediately apparent early in life but may be manifested in adulthood or during aging.
3. Importance of mixtures
If individuals and populations are exposed to an EDC, it is likely that other environmental pollutants are involved because contamination of environments is rarely due to a single compound. Furthermore, effects of different classes of EDCs may be additive or even synergistic (9).
4. Nontraditional dose-response dynamics
There are several properties of EDCs that have caused controversy. First, even infinitesimally low levels of exposure—indeed, any level of exposure at all—may cause endocrine or reproductive abnormalities, particularly if exposure occurs during a critical developmental window (10). Surprisingly, low doses may even exert more potent effects than higher doses. Second, EDCs may exert nontraditional dose-response curves, such as inverted-U or U-shaped curves (11). Both of these concepts have been known for hormone and neurotransmitter actions, but only in the past decade have they begun to be appreciated for EDCs.
5. Transgenerational, epigenetic effects
EDCs may affect not only the exposed individual but also the children and subsequent generations. Recent evidence suggests that the mechanism of transmission may in some cases involve the germline (12) and may be nongenomic. That is, effects may be transmitted not due to mutation of the DNA sequence, but rather through modifications to factors that regulate gene expression such as DNA methylation and histone acetylation.
B. The role of endocrinologists in discerning effects of EDCs
The field of endocrine disruption has particular pertinence to endocrinologists. In general, persistent endocrine disruptors have low water solubility and extremely high lipid solubility, leading to their bioaccumulation in adipose tissue. The properties of these substances are particularly well suited for study by endocrinologists because they so often activate or antagonize hormone receptors. There is no endocrine system that is immune to these substances, because of the shared properties of the chemicals and the similarities of the receptors (13) and enzymes involved in the synthesis, release, and degradation of hormones (Fig 1 1). ). Therefore, the role of this Scientific Statement is to provide perspectives on representative outcomes of exposures to endocrine disruptors and evidence for their effects in wildlife, laboratory animals, and humans.
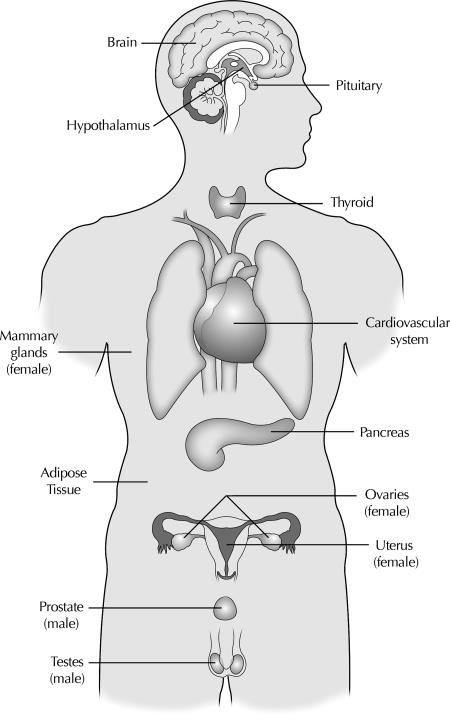
Model of the endocrine systems targeted by endocrine-disrupting chemicals as discussed in this article. This figure demonstrates that all hormone-sensitive physiological systems are vulnerable to EDCs, including brain and hypothalamic neuroendocrine systems; pituitary; thyroid; cardiovascular system; mammary gland; adipose tissue; pancreas; ovary and uterus in females; and testes and prostate in males.
II. Overview of Endocrine Disruption and Reproductive Health from a Clinical Perspective
A. Clinical aspects of endocrine disruption in humans
For a clinician taking care of an individual patient, there are numerous challenges in ascertaining EDC involvement in a particular disorder. Each person has unique exposure to a variety of both known and unknown EDCs. Individual differences in metabolism and body composition will create considerable variability in the half-life and persistence of EDCs, as well as their degradation in body fluids and tissues. Susceptibility to EDCs may vary according to genetic polymorphisms. In addition, human disorders are more likely the result of chronic exposure to low amounts of mixtures of EDCs. The latency between exposure to EDCs and occurrence of clinical disorders creates further challenges when one attempts to establish a relationship at the level of a given individual.
Epidemiological studies at the level of populations in a country or a region are crucial to alert researchers about geographical or secular trends in prevalence of disorders pointing to possible environmental factors. Registries with data on particular diseases or cell/organ donors may provide valuable contributions. For instance, the observation of adverse trends in male reproductive health together with declining sperm count in Denmark and other countries has led to the hypothesis of environmental contaminants being harmful to reproduction (14). Unfortunately, it is virtually impossible to make direct links between such epidemiological observations and exposure to given chemicals. Regional differences in certain reproductive disorders (infertility, cancer) that may be tied to contamination by compounds used locally such as in agriculture, industrial accident, or product misuse/abuse in subpopulations can also be informative (14,15). Finally, a comparison of disorders before and after migration to a new environment may reveal exposure and/or susceptibility to exposure to EDCs (16).
As already mentioned, a critical concern is the potential lag between exposure to EDCs and the manifestation of a clinical disorder. In humans, this period may be years or decades. In the case of reproduction, infertility cannot be assessed until the exposed individual has attained a certain age, again resulting in a lag between early exposure and manifestation of a dysfunction. Delayed or early puberty cannot be assessed until this event actually takes place, although timing of puberty could involve programming many years earlier during fetal life. Interestingly, an increased likelihood of early puberty was observed in subjects born with intrauterine growth retardation (IUGR) (17,18), suggesting a link between developmental programming and reproductive maturation. As discussed below, development of vaginal adenocarcinoma in women exposed fetally to DES (19) and the association of carcinoma in situ in the fetal testis with the development of testicular cancer in adulthood (14,20) are examples of links between the fetal environment and the occurrence of adult disease.
The timing of exposure is key to human disease because there are critical developmental periods during which there may be increased susceptibility to environmental endocrine disruptors. In those cases in which disruption is directed toward programming of a function, e.g., reproductive health, this may interfere with early life organization, followed by a latent period, after which the function becomes activated and the dysfunction can become obvious. For reproductive function in both humans and animals, fetal life is most vulnerable because there are rapid structural and functional events. The roles of sex steroids in sexual differentiation and thyroid hormones in brain development are of paramount importance at that time. Early postnatal life is also a time when maturation is still rapid (e.g., the central nervous system undergoes significant development at this time, including the hypothalamus which controls reproduction; see Section VII). The organization of the neuroendocrine control of reproduction is not completed at birth and remains sensitive to the interaction of steroids or EDCs neonatally such as has been shown for the control of ovulation in rodents. Breast or formula feeding could be of particular significance due to the capacity of human milk to concentrate EDCs in the former and the potential high intake of phytoestrogens in soy milk and/or plasticizers in formula-containing cans in the latter. It is apparent that the developmental basis of adult disease is an important concept for understanding endocrine disruption of reproductive function in humans.
B. Clinical dimorphism of EDCs on male and female reproduction
A spectrum of disorders throughout life, some of which are sexually dimorphic, can be related to endocrine disruption (Table 1 1). ). Male sexual differentiation is androgen-dependent (and potentially estrogen-dependent), whereas female differentiation occurs largely independently of estrogens and androgens. Therefore, it is expected that different disorders are seen in males and females as a result of EDC effects that overall mimic estrogens and/or antagonize androgens.
Table 1
Disorders of the human reproductive system possibly involving EDCs in their pathogenesis: A sexually dimorphic life cycle perspective
| Fetal/neonatal | Prepubertal | Pubertal | Adult | |
|---|---|---|---|---|
| Processes | Intrauterine growth | Adrenarche | Gonadarche | Spermatogenesis |
| Sexual differentiation | Ovulation | |||
| Hormonal control of prostate, breast, uterus, and lactation | ||||
| Male disorders | IUGR (15) | Premature pubarche | Small testes and high FSH (18) | Oligospermia (14,20) a |
| Cryptorchidism (14,20) a | Early puberty (25) | Testicular cancer (14,20) a | ||
| Hypospadias (14,20) a | Delayed puberty (25) | Prostate hyperplasia (24) | ||
| Female disorders | IUGR | Premature thelarche (25) | Secondary central precocious puberty (17,27) | Vaginal adenocarcinoma (19,28) |
| Peripheral precocious puberty (17) | PCOS (18,25) | Disorders of ovulation (29) | ||
| Premature pubarche (18) | Delayed ovulatory cycles (17,18) | Benign breast disease (29,31) | ||
| Breast cancer (30,31) | ||||
| Uterine fibroids (29) | ||||
| Disturbed lactation (29) |
a Cryptorchidism, hypospadias, oligospermia and testicular cancer are four components of the ″testicular dysgenesis syndrome″ as a common entity.
In the male (Table 2 2), ), cryptorchidism, hypospadias, oligospermia, and testicular cancer have been proposed to be linked as the testicular dysgenesis syndrome (TDS) arising from disturbed prenatal testicular development (14,21). Such links are important because they could mean that several disorders occur at different periods throughout life in a single individual as a result of exposure to a given EDC (or mixture) at a particular period. The epidemiological data relating TDS with environmental disruptors are indirect, and we still lack direct evidence of EDC involvement in the pathogenesis of TDS in humans (see Section V). In the rodent, however, a TDS-like condition can be observed after fetal exposure to phthalates (20), and the reduced anogenital distance observed in the rat (22) was observed in a recent epidemiological study on human male newborns (23). Several studies have shown a strong association of low birth weight with hypospadias and cryptorchidism, suggesting that they have a common determinant (15).
Table 2
Effects of some specific EDCs on the male reproductive system
| EDC | Exposed animal and effects | Possible translation to the clinical condition | Potential mechanisms |
|---|---|---|---|
| Vinclozolin | Fetal rat: hypospadias (36); undescended testes, prepubertal (37); delayed puberty (38), prostate disease among subsequent generations (34) | Epigenetic: altered DNA methylation in germ cell lines (12,34) | |
| DES | Fetal rats: hypospadias, cryptorchidism, micropenis, increased transmitted susceptibility to malignancies (28) | Hypospadias, cryptorchidism, micropenis, epididymal cysts (28) | Increased ERα expression in epididymis (43)) |
| Reduced insulin-like factor 3 (465 | |||
| DDT | Adult rats: decreased fertility (466) | Cryptorchidism | |
| DDE | Cryptorchidism | ||
| Phthalates | Reduced anogenital distance (22) | Reduced anogenital distance (23) and Leydig cell function, hypospadias | Decreased testosterone synthesis (468) |
| Cryptorchidism (467) | Cryptorchidism (14,20) | ||
| Oligospermia | Reduced fertility (14,20) | ||
| PCBs | Fetal rat: decreased spermatogenesis, delayed puberty | Reduced penile length, delayed sexual maturation, reduced fertility | |
| Fetal: testis cancer | |||
| BPA | Increased prostate size (469) | Increased ERα expression in hypothalamus (42) | |
| Aberrant development of prostate and urethra (470) | Increased AR expression in prostate (469) | ||
| Prostate cancer (122) | |||
| Increased anogenital distanceAltered periductal stroma in the prostate (471) |
Other pathologies in males are linked to EDC exposure. Prostate hyperplasia has been described after exposure to BPA (24). In adolescence, boys born with IUGR have small testes and elevated serum FSH, together with low inhibin B levels (18) that could be related to some of the TDS disorders. Divergent data have been reported on effects of EDCs on pubertal timing in the male (25).
In the female (Table 3 3), ), premature thelarche has been reported in girls exposed to phthalates (26), although these data need to be replicated. Sexual precocity presumably of peripheral origin initially and secondarily central could be related to exposure to the insecticide DDT in girls migrating for international adoption (17). A neuroendocrine mechanism is suggested by experiments in a rodent model (27) (see Section VII). An association of premature pubarche and ovulatory disorders with EDCs is suggested indirectly by links with IUGR at birth and metabolic syndrome in adulthood (18).
Table 3
Effects of some specific EDCs on the female reproductive system
| EDC | Exposed animal and effects | Possible translation to the clinical condition | Potential mechanisms |
|---|---|---|---|
| Vinclozolin | Fetal rat: multisystem disorders including tumors (12) | Epigenetic: altered DNA methylation in germ cell line (12); reduced ERα expression in uterus (44) | |
| DES | Fetal mouse: transmitted susceptibility to malignancies (39) | Vaginal adenocarcinoma in daughters of women treated with DES during pregnancy (19) | |
| DDT/DDE | Immature female rat: sexual precocity (27) | Precocious and early puberty (17) | Neuroendocrine effect through estrogen receptors, kainate receptors, and AhRs (27) |
| Reduced fertility in daughters of exposed women (472) | |||
| BPA | Inhibited mammary duct development and increased branching (145) | Miscarriages | Inhibition of apoptotic activity in breast (145);0>Increased number of progesterone receptor-positive epithelial cells;0>Reduced sulfotransferase inactivation of estradiol (45,46);0>Nongenomic activation of ERK1/2 (476) |
| Increased mammary gland density, increased number of terminal ends (146) | |||
| Reduced weight of vagina (473) | |||
| Endometrial stimulation (473) | |||
| Early puberty (474,475) | |||
| PCBs | Fetal and early postnatal rat: neuroendocrine effects in two generations, and behavioral changes (296,477) | Actions on estrogen receptors, neurotransmitter receptors | |
| Dioxins | Fetal rat: altered breast development and increased susceptibility for mammary cancer (478) | Inhibition of cyclooxygenase2 via AhR (479) | |
| Early pubertal rat: blocked ovulation | |||
| Phthalates | Premature thelarche (25) |
In the adult female, the first evidence of endocrine disruption was provided almost 40 yr ago through observations of uncommon vaginal adenocarcinoma in daughters born 15–22 yr earlier to women treated with the potent synthetic estrogen DES during pregnancy (19). Subsequently, DES effects and mechanisms have been substantiated in animal models (28). Thus, robust clinical observations together with experimental data support the causal role of DES in female reproductive disorders. However, the link between disorders such as premature pubarche and EDCs is so far indirect and weak, based on epidemiological association with both IUGR and ovulatory disorders. The implications of EDCs have been proposed in other disorders of the female reproductive system, including disorders of ovulation and lactation, benign breast disease, breast cancer, endometriosis, and uterine fibroids (29,30,31,32).
C. Experimental and clinical evidence of EDCs and potential mechanisms
In Tables 2 2 and 3 3, , some experimental and clinical observations of disturbed reproductive systems are listed for selected EDCs. The evidence from human epidemiological studies is partial and indirect (see Section V). Mechanistic studies are ethically and practically very limited in humans and have to rely on data obtained using animal experiments (in vivo and in vitro models), although these models can have limitations. Clinical and experimental studies correlate DES effects quite convincingly in both sexes. In the male, rodent studies using phthalates and, to a lesser extent, PCBs model TDS entirely or partly. In the female, some rodent studies are consistent with DDT/dichlorodiphenyldichloroethylene (DDE) involvement in sexual precocity.
The following considerations emphasize some of the concepts emerging from the available data.
1. Heritability
There may be transgenerational effects of EDCs due to overt mutation or to more subtle modifications of gene expression independent of mutation (i.e., epigenetic effects). Epigenetic effects of EDCs include context-dependent transmission (e.g., the causal factor persists across generations; Ref. 33) or germline-dependent mechanisms (i.e., the germline itself is affected; Refs. 12, 34, and 35). An example of germline transmission of an epigenetically modified trait is shown in a rat model for the fungicide vinclozolin and is manifested by a higher likelihood of metabolic disorders, tumors, and reproductive dysfunctions in the next four generations (12,34,35,36,37,38). In the case of DES, there are both human and experimental observations indicating heritability (19,28,39).
2. Diversity and complexity of mechanisms
EDCs often act via more than one mechanism. Some EDCs have mixed steroidal properties: for example, a single EDC may be both estrogenic and antiandrogenic. EDCs may be broken down or metabolized to generate subproducts with different properties. For instance, the estrogen agonist DDT is metabolized into the androgen antagonist DDE (27). The balance between estrogenic and androgenic properties of EDCs can be biologically significant because reproduction of both sexes involves an interplay of androgens and estrogens. In humans, early breast development occurs in girls with a highly active variant of CYP3A4, a cytochrome p450 enzyme involved in inactivating testosterone (40), and premature thelarche occurs with antiandrogenic phthalates (25). Similar androgen-estrogen interactions have been reported in DES-treated rats in which reduced androgen secretion or action sensitized the animals to the estrogenic effects of DES (41). Moreover, many organs are targeted by sex steroids and are thereby vulnerable to endocrine disruption, including the hypothalamic-pituitary-gonadal system, breast, uterus, cervix, vagina, brain, and nonreproductive tissues such as bone, muscle, and skin (Fig. 1 1). ). In the case of humans, a peripheral effect in the reproductive system (e.g., breast development) can result from direct EDC effects (peripheral puberty) and/or endogenous estrogen increase through premature neuroendocrine maturation (central puberty) (17,27), but these may be difficult to distinguish. For instance, EDC effects can involve altered ERα expression in hypothalamus (42) and epididymis (43) or uterus (44). Along with the direct influence of EDCs on estrogen or androgen actions, they can affect endogenous steroid production through negative and positive feedback, effects that may differ depending on developmental stage. Also, there are multiple levels of interactions with steroid action (receptor or promoters), synthesis (e.g., aromatase stimulation by atrazine), and metabolism [e.g., sulfotransferase (45)]. Finally, there are coexisting mechanisms not directly mediated at the hypothalamic-pituitary-gonadal (HPG) system. For instance, reproductive dysfunction can result from thyroid disruption (46) or nonspecific interference of reduced energy intake (47).
3. Limits of translational models
The in vivo animal models may be difficult to extrapolate to humans for several reasons, including species differences in ontogeny of reproductive system and functions, differences in metabolism of sex steroids, difficulty in estimating exposure to mixtures, and variable body burdens. As already mentioned, exposure to EDCs is complex. For example, mixtures are likely to be the usual form of exposure to EDCs, but they are difficult to approximate in experimental models. Moreover, the effects may not be additive; nevertheless, a combination of low doses of substances that individually are inactive may result in a biological perturbation (48). Despite these limitations, considering the substantial conservation of endocrine and reproductive processes across species, it is certainly reasonable to use animal models for understanding human processes, as long as these potential differences are taken into account.
III. Clinical and Translational Impacts of EDCs on Female Reproduction
A. Introduction to female reproductive development and function
Development and function of the female reproductive tract depends on coordinated biological processes that, if altered by endogenous or exogenous factors during critical periods of development or during different life stage, could have significantly adverse effects on women’s health and reproductive function and outcomes. For example, the full complement of cell types in the human ovary depends on successful germ cell migration from the yolk sac during the first trimester and differentiation into oocytes with associated somatic cells to form the functional unit of the primordial follicle by the second to third trimesters of gestation. Factors that interfere with germ cell migration or follicle formation can result in abnormal functioning of this tissue with significant reproductive consequences. Also, the oocyte is arrested in the diplotene stage of late prophase until meitic divisions occur beginning at puberty (meiosis I) and after fertilization (meiosis II), and abnormalities in these processes can have a profound impact on reproductive outcomes, such as aneuploidy, premature ovarian failure (POF), and miscarriage. In addition, whereas Mullerian tract formation begins at 8 wk gestation with fusion of the Mullerian ducts and subsequent differentiation into the uterus (endometrium, myometrium), cervix, and upper vagina, uterine differentiation with regard to formation of luminal epithelium, glandular epithelium, and stromal components is mostly a postnatal event, with functionality of response to steroid hormones beginning at puberty. Interference with these processes can predispose women to infertility, ectopic gestation, poor pregnancy outcomes, and other reproductive disorders that may be programmed during development (e.g., endometriosis, uterine fibroids). Thus, abnormal development or alterations at other times in the life cycle can alter anatomy and functionality of the female reproductive tract and thus can alter the reproductive potential of affected individuals and their offspring.
Most female reproductive disorders are well described with regard to clinical presentation, histological evaluation of involved tissues where applicable, and diagnostic classification. However, whereas few are polygenic inherited traits and some are due to infections, the pathogenesis of the vast majority of female reproductive disorders is not well understood. This has hindered a preventive strategy to their development and/or exacerbation, and in some cases limited the development of effective therapies for symptoms and associated morbidities.
A key question arises as to whether EDCs contribute to the development of female reproductive disorders, particularly those occurring during a critical window of susceptibility: in utero, neonatally, in childhood, during puberty, and during adulthood. There are increasing data from wildlife studies and laboratory studies with rodents, ungulates, and nonhuman primates that support a role of EDCs in the pathogenesis of several female reproductive disorders, including polycystic ovarian syndrome, aneuploidy, POF, reproductive tract anomalies, uterine fibroids, endometriosis, and ectopic gestation (for reviews, see Refs. 29 and 49,50,51,52,53,54; also see Table 4 4). ). Many of the mechanisms are understood and, moreover, are conserved between animals and humans. Herein, we describe some of the clinical implications of these associations.
Table 4
Female reproductive disorders and their possible relationships to EDCs: Some experimental and human data
| Female reproductive disorder | Experimental data | Human epidemiological data |
|---|---|---|
| Reproductive tract abnormalities/ malignancies | Mice prenatally exposed to DES have structural abnormalities of the oviduct, uterus, cervix, and vagina, leiomyoma, infertility-subfertility, immune dysfunction, ovarian cysts, ovarian tumors, vaginal adenocarcinoma (480) | In utero exposure to DES: abnormal cervical, uterine, and oviduct anatomy (481), vaginal adenocarcinoma (19), subfertility and infertility, ectopic pregnancy (480) |
| Endometriosis | Adult monkey exposed to TCDD (dioxin): promotion of growth and survival of endometriosis impants (110) | ↑ plasma concentrations of DEHP in women with endometriosis vs. controls (113); ↑ levels of phthalates (DnBP, BBP, DnOP, DEHP) in Indian women with endometriosis vs. controls (114) |
| Precocious puberty | Immature female rat exposed to DDT: sexual precocity (27) | High levels of the DDT metabolite p,p′-DDE, in plasma from foreign immigrant girls with precocious puberty in Belgium (482) |
| Female mouse fetuses exposed to BPA: early puberty (474) | Breastfed girls exposed to high levels of PBB in utero (≥7 ppm): earlier age at menarche (483) | |
| Premature thelarche | Higher levels of phthalates and its major metabolite mono-(2-ethylhexyl) phthalate in serum of girls from Puerto Rico with premature breast development (26) | |
| Disturbed lactation | Rodents exposed to atrazine: impaired lactation through prolactin inhibition (484) | Negative correlation between DDE (metabolic product of DDT) and duration of lactation (484) |
| Breast abnormalities/cancer | Fetal rats exposed to dioxins (TCDD): altered breast development and ↑ susceptibility for mammary cancer (478) | Limited and conflicting evidence |
| Mice exposed to BPA: altered organization of the mammary anlagen, accelerated ductal development, and inhibition of lumen formation in the fetus (128) | M2 polymorphism in the cytochrome P450 1A1 gene modify the association between PCB exposure and risk of breast cancer (51) | |
| Mice exposed to BPA: increased number of epithelial structures (145,146) | ||
| Rats exposed perinatally to BPA: development of preneoplastic lesions (intraductal hyperplasias) and carcinomas in situ (148) | ||
| Rats exposed perinatally to BPA; increased susceptibility to neoplastic development (149) | ||
| Rats: lactational exposure to BPA: shortening of the latency period and increased tumor multiplicity after carcinogen challenge (150) | ||
| Mice exposed to BPA: development of preneoplastic lesions (intraductal hyperplasias) (147) | ||
| PCOS | Prenatal exposure to high levels of testosterone results in fetal programming of PCOS traits (60,61) | Increased levels of serum AGEs in women with PCOS and positive correlation between AGE proteins and testosterone levels (64) |
| Rats fed with high vs. low AGE diet: ↑ androgens–↑ ovarian volume and AGE ovarian deposition (461) | In polycystic ovaries, increased immunostaining of colocalized AGEs, RAGEs, and activated nuclear factor-κB (211,485) | |
| Fertility and fecundity | Mice prenatally exposed to DES (480) | Isolation of persistent organochlorine chemicals from ovarian follicular fluid of women undergoing IVF (51) |
| Indications that exposure to pesticides may contribute to female infertility in some occupationally exposed groups (484) |
↑, Increased; DEHP, di-(2-ethylhexyl) phthalate; DnBP, di-n-butyl phthalate; BBP, butyl benzyl phthalate; DnOP, di-n-octyl phthalate.
B. Polycystic ovarian syndrome (PCOS)
PCOS is a heterogeneous syndrome characterized by persistent anovulation, oligo- or amenorrhea, and hyperandrogenism in the absence of thyroid, pituitary, and/or adrenal disease (55,56,57). At the level of the ovary, there is recruitment and growth of follicles to the small antral stage, without selection of a dominant, preovulatory follicle, leading to accumulation of multiple, small, antral follicles (58). Hyperfunctioning of the theca and relative hypofunctioning of the granulosa cells accompany the acyclicity of the syndrome. Many, but not all women with PCOS have relatively high circulating levels of LH, compared with FSH, believed to be due to insensitivity to steroid hormone feedback. However, this does not fully account for the observed increase in thecal androgen production or the relative quiescence and sometimes frank FSH resistance of the granulosa cells. This complex disorder likely has its origins both within and outside the hypothalamic-pituitary-ovarian axis, and metabolic, neuroendocrine, and other endocrine regulators likely contribute to its manifestation. Obesity and insulin resistance occur in about 50% of women with PCOS, and obese women have a 12% risk of having PCOS (59). PCOS has multiple physiological processes (e.g., neuroendocrine functioning and feedback mechanisms, ovarian steroidogenesis, insulin resistance, and obesity) that are regulated by hormonal and metabolic parameters. Hence, endocrine disruption by environmental chemicals may indeed contribute to the pathogenesis of PCOS.
In sheep and rhesus monkeys, prenatal exposure to high levels of testosterone results in fetal programming of PCOS traits (60). Specifically, high levels of testosterone exposure at gestational d 40–60 and 100–115 result in rhesus monkey females who, in adulthood, have anovulatory infertility, hypersecretion of LH, elevated circulating levels of testosterone, neuroendocrine feedback defects, central adiposity and compensatory insulin resistance, and polycystic ovaries with ovarian hyperandrogenism and follicular arrest in adulthood (60,61). In the sheep model, a similar PCOS phenotype, along with IUGR and compensatory catch-up growth after birth, derives from prenatal exposure to exogenous testosterone (60,62). In rhesus monkey and sheep, unlike rodents, follicular differentiation is completed during fetal life. Thus, it is plausible that in utero exposure of human female fetuses to androgen-like EDCs could result in PCOS in adulthood, along with associated metabolic disorders. Very recent evidence for androgenic properties of personal-care products such as triclocarban (63) add to the possibility of environmental androgens, although a connection to PCOS has not yet been drawn.
There are numerous candidate genes associated with predisposition to developing PCOS in women (57,64), and how and if these interact with prenatal androgen-like factors to promote the PCOS phenotype in women has not been determined. Nonetheless, PCOS is a debilitating disorder in women, occurring in 6.6% of the reproductive-age population (65,66,67); it is a leading cause of subfertility and is associated with increased lifetime risks for cardiovascular disease and type II diabetes (55). In addition to these clinical impacts on patients, the cost to the health care system for PCOS diagnosis and treatment is substantial, totaling in 2004 about $4.4 billion in the United States alone (68). These facts underscore the need to understand potential EDC contributions to the development of PCOS in an effort to minimize such exposures and maximize prevention.
Other pathways may be involved in endocrine disruption of PCOS. Women with PCOS have higher levels of the EDC BPA (69), and increased testosterone in these women is consistent with decreased clearance of BPA (70). Although adult exposures do not necessarily imply earlier exposures in life, especially with EDCs of relatively short half-lives, there are data demonstrating nearly 5-fold higher levels of BPA in amniotic fluid compared with other body fluids, suggesting significant prenatal exposure (71). Although a cause and effect of BPA and PCOS have not been demonstrated definitively, the biological plausibility is interesting and worthy of further consideration.
C. Premature ovarian failure, decreased ovarian reserve, aneuploidy, granulosa steroidogenesis
POF (cessation of proper ovarian function before the age of 40) occurs in about 1% of reproductive-age women (72). Although in some cases the causation is known, for the vast majority of women with POF this is not the case, and there are stages of susceptibility during organogenesis and adult exposures that could contribute to POF.
Because the total ovarian follicle complement is established before birth in humans (73), anything that interferes with this, resulting in a decreased ovarian follicle resting pool, can result in POF. For example, disruption of germ cell migration from the genital ridge into the developing gonad results in ovarian dysgenesis. The resting pool undergoes a baseline level of apoptosis, and TNF-α, Fas ligand, and androgens stimulate this in the resting pool, as well as in the growing pool (74). Also, once a cohort of follicles is recruited during a given cycle in women, survival factors (FSH, estradiol, and growth factors, e.g., IGFs) are important for escape from apoptosis of the dominant follicle. Recent data in the mouse show that selective activation of the K-ras pathway in the oocyte results in rapid follicular development and depletion (75). Interestingly, adult and in utero exposures of mice to BPA have resulted in damage to oocytes (76,77). Specifically, adult exposures result in abnormalities in alignment of chromosomes on the meiotic spindle and aneuploidy, which, while not leading to ovarian senescence, does lead to aneuploid gametes and offspring (76). However, BPA given to pregnant dams during midgestation affects the developing ovary with resulting abnormalities in meiotic prophase, including synaptic defects, and mature animals exposed in utero have an increase in aneuploid oocytes and embryos (77). Such alterations also lead to cell cycle arrest and oocyte death, thus depleting the complement of normal oocytes (77). Currently, there are no data on in utero or adult exposure to BPA and aneuploidy in humans, but the possibility that there are parallels is compelling.
Interestingly, mice exposed in utero to DES, between d 9–16 gestation, have a dose-dependent decrease in reproductive capacity, including decreased numbers of litters and litter size and decreased numbers of oocytes (30%) ovulated in response to gonadotropin stimulation with all oocytes degenerating in the DES-exposed group, as well as numerous reproductive tract anatomic abnormalities (78). In women with in utero exposure to DES, Hatch et al. (79) reported an earlier age of menopause between the 43–55 yr olds, and the average age of menopause was 52.2 yr in unexposed women and 51.5 yr in exposed women. The effect of DES increased with cumulative doses and was highest in a cohort of highest in utero exposure during the 1950s (79). These observations are consistent with a smaller follicle pool and fewer oocytes ovulated, as in DES-exposed mice after ovulation induction (78).
Of interest are human data that demonstrate unequivocally that adult exposure in women to cigarette smoke results in decreased fecundity, decreased success rates in in vitro fertilization (IVF), decreased ovarian reserve (higher basal cycle d 3 FSH and stimulated parameters), earlier menopause by 1–4 yr, and an increased miscarriage rate (80,81). The mechanism appears to be mediated by the AhR-mediated apoptosis of oocytes, with accelerated loss of ovarian follicles. Interestingly, exposure of rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in utero and through the end of reproductive life results in a dose-dependent onset of premature reproductive senescence, likely due to direct effects on ovarian function (82).
Thus, whereas POF may occur in a relatively small percentage of the population, there are several alarming signals that should not be ignored. For example, the age group with the fastest growing rate of involuntary subfertility is 15- to 24-yr-old women (83). Also, the known effects of environmental contaminants on oocyte survival, aneuploidy, decreased ovarian reserve, and infertility described above underscore how much at risk the population may be for reproductive compromise.
With regard to ovarian granulosa steroidogenesis, several EDCs have effects on this process (84). For example, TCDD (10 ppm) decreases FSH-stimulated LH receptor mRNA expression and half-life in cultured granulosa (85). DDE increases vascular endothelial growth factor and IGF-I expression in luteinized granulosa from IVF patients, suggesting a contribution to impaired steroidogenesis and perhaps infertility (86). Recently, Kwintkiewicz and Giudice (87,88) have shown, in preliminary studies, that BPA decreases proliferation and FSH-induced aromatase expression via activation of peroxisome proliferator-activated receptor γ (PPAR-γ) and increases IGF-I and IGF receptor type I in human granulosa-like tumor cells and luteinized human granulosa from IVF subjects. These data suggest that EDCs may have local effects on ovarian function in adult women.
D. Reproductive tract anomalies
Disruption of female reproductive tract development by the EDC DES is well documented (89). A characteristic T-shaped uterus, abnormal oviductal anatomy and function, and abnormal cervical anatomy are characteristic of this in utero exposure, observed in adulthood (90), as well as in female fetuses and neonates exposed in utero to DES (91). Some of these effects are believed to occur through ERα (92) and abnormal regulation of Hox genes (93,94). Clinically, an increased risk of ectopic pregnancy, preterm delivery, miscarriage, and infertility all point to the devastating effect an endocrine disruptor may have on female fertility and reproductive health (89). It is certainly plausible that other EDCs with similar actions as DES could result in some cases of unexplained infertility, ectopic pregnancies, miscarriages, and premature deliveries. Although another major health consequence of DES exposure in utero was development of rare vaginal cancer in DES daughters, this may be an extreme response to the dosage of DES or specific to pathways activated by DES itself. Other EDCs may not result in these effects, although they may contribute to the fertility and pregnancy compromises cited above. Of utmost importance clinically is the awareness of DES exposure (and perhaps other EDC exposures) and appropriate physical exam, possible colposcopy of the vagina/cervix, cervical and vaginal cytology annually, and careful monitoring for fertility potential and during pregnancy for ectopic gestation and preterm delivery (89,95).
E. Uterine leiomyomas
Uterine leiomyomas (fibroids) are benign smooth muscle tumors of the myometrium that can cause morbidity for women, including menorrhagia, abdominal pain, pelvic prolapse, and infertility and miscarriage (96). They are the most common tumor of the reproductive tract in women and comprise the leading cause for hysterectomy and the second leading cause of inpatient surgery in the United States, with health care costs exceeding $2 billion in 2004 (97). The prevalence rate of uterine leiomyomas is approximately 25–50%, with a preponderance occurring in African-American women (97). The greatest risk factor in adult women is prolonged exposure to unopposed estrogen. Whether in utero exposure to DES increases a woman’s lifetime risk of developing uterine fibroids is controversial, as the method to detect fibroids in two different studies influenced the outcome (98,99). Specifically, in a study of 1731 women exposed to DES and 848 matched unexposed controls, no association was found (P = 0.68) when histological confirmation after myomectomy or hysterectomy was used to document uterine fibroids (98). In contrast, when ultrasound was used to determine the presence of fibroids in DES-exposed vs. DES-unexposed women, a significant relationship was found (odds ratio, 2.4; 95% confidence interval, 1.1–5.4) in DES-exposed women and uterine fibroids (99). However, there are strong animal data to support development of uterine fibroids in adulthood after in utero exposure to EDCs, especially DES (for reviews, see Refs. 49, 50, and 52). Newbold et al. (100) reported that CD-1 mice develop uterine leiomyomas if exposed in utero or neonatally to DES, whereas unexposed mice do not. Furthermore, the Eker rat, which has a germ-line mutation in the rat homolog of the tuberus sclerosis complex 2 tumor suppressor gene, spontaneously develops uterine leiomyomas (101). The number, size, and growth rate of the fibroids increase significantly when the rat is exposed to DES on postnatal d 3–5 and 10–12, but not 17–19 (102), an effect that can be diminished with prior oophorectomy (102). These data overall strongly suggest developmental programming and gene-environment interactions for the increased risk of uterine lyomyomas in this rat model (103). In addition to mice, the Eker rat, and some dogs, the Baltic gray seal that has high organochlorine body burden also develops uterine leiomyomas (104). As with most environmental causes of abnormalities in the reproductive tract (and other tissues and organs), direct cause and effect relationships are difficult to establish. However, as in many of the other abnormalities in this Scientific Statement, the likelihood of such a relationship is plausible.
F. Endometriosis
Endometriosis is an estrogen-dependent gynecological disorder associated with pelvic pain and infertility. It occurs in 6–10% of women and up to 50% of women with pelvic pain and infertility. In 2002, the total health care costs estimated in the United States for diagnosis and treatment of endometriosis totaled approximately $22 billion (105). There are suggestive animal data of adult exposure to EDCs and development of or exacerbation of existing disease, and there is evidence that in utero exposure in humans to DES results in an increased relative risk = 1.9 (95% confidence interval, 1.2–2.8) (106). Most striking are the observations of rhesus monkeys administered different doses of TCDD and their subsequent development of endometriosis (107,108). Although this study had low sample size and confounding variables that brought into question the relationship between endometriosis and TCDD (49,52,109), another study revealed that adult exposure of cynomolgus monkey to TCDD promotes growth and survival of endometriosis implants (110), indicating that this EDC is involved in the progression, if not pathogenesis, of this disorder. Similar data were obtained in rodent models of endometriosis in which human endometrium is transplanted into mouse and rat peritoneum, and the established lesions grew larger when animals were exposed to TCDD in utero and as adults (111,112), underscoring the estrogen (and EDC) dependence of this disorder.
There are also correlative findings of phthalate levels in plasma and endometriosis. For example, Cobellis et al. (113) found high plasma concentrations of di-(2-ethylhexyl)-phthalate in women with endometriosis, and an association of phthalate esters with endometriosis was found among Indian women (114). Thus, the evidence is accumulating of correlations between EDCs in the circulation of women with endometriosis, although a cause-and-effect relationship has yet to be established, which is not uncommon in reproductive environmental toxicity.
Endometriosis is believed to be due to retrograde menstruation and transplantation of endometrial fragments and cells into the peritoneal cavity. Because nearly all women have retrograde menstruation but relatively few have endometriosis, the disorder is also believed to involve a dysfunctional immune response, i.e., activated macrophages in the peritoneal cavity with robust secretion of inflammatory cytokines but without clearance of disease. An interesting model of early-life immune insult and developmental immunotoxicity suggests that in utero exposures to specific insults may reprogram the immune system, resulting in disorders such as chronic fatigue syndrome, cancer, and autoimmune disorders. Whether this has any relevance to the development or progression of endometriosis in adult women has not been explored but warrants further evaluation. Interestingly, TCDD and a therapy for endometriosis, danazol, both have effects on the adult immune system, although effects on the developing immune system are not known.
Although the infertility associated with endometriosis for the most part can be treated with advanced reproductive technologies, less success has been achieved with treatment of endometriosis-related pain. Because the pathogenesis of the associated pain is not known with certainty, therapies are empiric and include agents directed to minimize inflammation (nonsteroidal antiinflammatory drugs, danazol), progestins and androgens (to oppose estrogen actions), GnRH analogs (to inhibit gonadotropin secretion and thus ovarian estradiol production), and aromatase inhibitors (to inhibit estradiol synthesis by the ovary and endometriotic lesions), as well as surgical ablation or excision of the disease, when possible. Most of these therapies are effective in up to 50–60% of affected women, with either intolerable side effects (e.g., profound hypoestrogenism) or recurrence of pain (e.g., after surgery) (115). Thus, prevention is key to this disorder, as is understanding the pathogenesis so that therapies for pain can be devised appropriately and administered.
IV. Endocrine Disruptors, Mammary Gland Development, and Breast Cancer
It has been hypothesized that the significant increase of the incidence of breast cancer in the industrialized world observed during the last 50 yr may be due to exposure to hormonally active chemicals, particularly xenoestrogens (116). A similar increase in the incidence of testicular cancer and malformations of the male genital tract and decreased quantity and quality of human sperm have been observed during the same half century, again suggesting a link to the introduction of these chemicals into the environment (117) (see Sections II and V).
A. Windows of vulnerability to carcinogenic agents and “natural” risk factors
The standard risk factors for developing breast cancer include age at menarche, first pregnancy, menopause, lactation, and parity. All of these factors are related to lifetime exposures to ovarian hormones. It is also known that there are developmental periods of enhanced vulnerability (see Section I). For example, sensitivity to radiation is highest during puberty. Additionally, pregnancy increases the risk of breast cancer in the short term (118) and decreases it in the long term (119). More recently, epidemiological studies have revealed that the intrauterine environment may also influence the risk to develop breast cancer later in life. Studies comparing human dizygotic twins and single births revealed that the propensity to breast cancer is enhanced in female twins, and this outcome was attributed to excess estrogen exposure in dizygotic twins during gestation (120).
B. Theories of carcinogenesis
A majority of researchers support the idea that cancer is due to the accumulation of mutations in a cell [the somatic mutation theory (121)]. In contrast, supporters of the theory of developmental origins of adult disease are proposing that changes in the epigenome play a central role in carcinogenesis (see Section VI).
Both the genetic and epigenetic theories of carcinogenesis imply that cancer originates in a cell that has undergone genetic and/or epigenetic changes, which ultimately results in dysregulated cell proliferation (122). Alternatively, the tissue organization field theory postulates that carcinogenesis represents a problem of tissue organization, comparable to organogenesis gone awry, and that proliferation is the default state of all cells (123,124,125). According to this theory, carcinogens, as well as teratogens, would disrupt the normal dynamic interaction of neighboring cells and tissues during early development and throughout adulthood (126).
During postnatal life, the mammary gland undergoes massive architectural changes, comparable to those usually associated with organogenesis. These changes occur in response to alterations in endogenous hormone levels such as those associated with puberty and pregnancy and can be induced experimentally by endocrine manipulation. Many studies of endocrine disruptors have illustrated that developmental exposure to these exogenous hormone mimics can alter normal patterns of tissue organization and hence disrupt stromal-epithelial interactions (127,128). These changes may disturb important regulatory mechanisms and enhance the potential for neoplastic lesions.
C. Susceptibility of the breast during puberty and adulthood
Several epidemiological studies explored the link between exposure to endocrine disruptors and breast cancer incidence. In general, these are case-control studies that usually measure exposure to a single chemical at the time of breast cancer diagnosis. This type of study has produced inconsistent results. Prospective studies that measured exposure several years before cancer diagnosis revealed a positive link between breast cancer and chemical exposure to toxaphene (129) and DDT (130). In particular, a study linked DDT with an increased risk of breast cancer when the exposure was measured before 14 yr of age. This study used samples taken before the banning of DDT for agricultural use and hence represents higher exposures than those measured today. Humans, however, are exposed to a plethora of hormonally active chemicals with different metabolic profiles. Moreover, individuals living in the same area may be exposed to a different mixture of chemicals due to different diets and to migration history. These facts imply that a single chemical cannot be construed as a marker of total exposure. Not surprisingly, one case-control study reported a significant correlation between total xenoestrogen exposure and breast cancer (131).
How xenoestrogen exposure during the period of sexual maturity may result in mammary gland carcinogenesis remains unsolved; this is not surprising because the mechanisms underlying hormonal carcinogenesis are still unknown. One possibility, compatible with all the cancer hypotheses briefly discussed above, is that xenoestrogens may extend the length of the period of ductal growth and alveologenesis during the menstrual cycle. This period is also characterized by proliferative activity in the glandular epithelium. For example, ductal cell proliferation in the breast is maximal from the late follicular phase and throughout the luteal phase, i.e., when endogenous estrogen levels are high (132). The ubiquitous presence of xenoestrogens in foods, their persistence, and their lack of binding to the plasma carrier protein SHBG (127) may result in relatively constant levels in blood. These xenoestrogens would act additively with ovarian estrogens and thus advance by a few days the period of ductal growth. Hence, a small and maintained increase of estrogenic activity during the period of low ovarian output could be sufficient to “promote” carcinogenesis by increasing the number of cells that undergo proliferation menstrual cycle after menstrual cycle, an explanation consistent with the somatic mutation theory. An alternative explanation, consistent with the tissue organization field theory, is that estrogens acting as morphogens would enhance tissue remodeling through stroma epithelium interactions and increase the likelihood of producing alterations of tissue architecture. This notion is supported by data showing that recombination of normal mammary epithelial cells with stroma exposed to carcinogenic agents results in the development of epithelial neoplasias (133) and that conversely, recombination of mammary carcinoma cells with stroma from multiparous animals (which are refractory to carcinogens) results in the normalization of the neoplastic phenotype (126).
D. Susceptibility of the mammary gland during the perinatal period
Direct evidence of prenatal estrogen exposure and breast cancer risk is being gathered from the cohort of women born to mothers treated with DES during pregnancy and is discussed above (see Sections II and III). These women are now reaching the age at which breast cancer becomes more prevalent. In the cohort of these women who are aged 40 yr and older, there is a 2.5-fold increase in the incidence of breast cancer compared with unexposed women of the same age (134,135), suggesting that indeed, prenatal exposure to synthetic estrogens may play an important role in the development of breast neoplasms. Consistent with this, experiments in rats showed that prenatal exposure to DES resulted in increased mammary cancer incidence during adulthood (136,137). These experiments illustrated that rats exposed prenatally to DES and challenged with the chemical carcinogen dimethylbenzanthracene (DMBA) at puberty had a significantly greater incidence of palpable mammary tumors at 10 months of age than animals exposed prenatally to vehicle. In addition, the tumor latency period was shorter in the DES-exposed compared with the vehicle-exposed group (130). Both the epidemiological and experimental data are consistent with the hypothesis that excessive estrogen exposure during development may increase the risk of developing breast cancer.
In utero exposure to tamoxifen, an estrogen antagonist and partial agonist, has also been shown to increase the incidence of mammary tumors when the exposed offspring are challenged with DMBA at puberty. Eighteen weeks after the challenge, 95% of the tamoxifen-exposed animals developed tumors, compared with 50% of the vehicle-treated rats (138). However, in the above-mentioned studies, both DES and tamoxifen were administered at high pharmacological doses to reflect the medical use of these agents, whereas the effects of twinning mentioned above represent a physiological range of endogenous hormone levels to which developing fetuses are exposed.
E. Perinatal exposure to environmentally relevant levels of endocrine disruptors
There is a third type of exposure that needs to be addressed: the inadvertent and continuous exposure of fetuses to environmentally active chemicals, such as dioxins and BPA (Table 4 4 ).
1. Dioxins
Depending on the context (time of exposure, organ, presence or absence of estrogens) dioxins have either estrogenic or antiestrogenic effects. Despite cross-talk between the aryl hydrocarbon and ERs (139), the mechanisms underlying these opposite effects have yet to be elucidated. Rats exposed prenatally (gestational d 15) to TCDD and challenged with the chemical carcinogen DMBA at 50 d of age showed increased tumor incidence, increased number of tumors per animal, and shorter latency period than rats exposed prenatally to vehicle and to DMBA at 50 d of age. These TCDD-exposed animals had increased numbers of terminal end buds at puberty (140). Because these structures are believed to be the site where mammary cancer arises, these results were interpreted as evidence that TCDD increased the propensity to cancer by altering mammary gland morphogenesis. Interestingly, Fenton (31) showed that prenatal exposure to TCDD results in impaired development of terminal end buds that remain in the gland for prolonged periods, whereas in the normal animals terminal end buds are transient structures that regress when ductal development is completed.
2. BPA, a ubiquitous xenoestrogen
The ubiquitous use of BPA provides great potential for exposure of both the developing fetus, indirectly through maternal exposure, and the neonate, directly through ingestion of tinned food, infant formula, or maternal milk (11). Indeed, BPA has been measured in maternal and fetal plasma and placental tissue at birth in humans (141). A recently published study conducted by the Centers for Disease Control, the first using a reference human population, showed that 92.6% of over 2500 Americans had BPA in their urine (142). Measured urine concentrations were significantly higher in children and adolescents compared with adults. BPA has also been measured in the milk of lactating mothers. These data indicate that the developing human fetus and neonate are readily exposed to this chemical.
In rodents, BPA has been shown to readily cross the placenta (143,144) and bind α-fetoprotein (the estrogen-binding protein that prevents maternal estrogen from entering the circulation of the fetus) with negligible affinity relative to estradiol; this results in enhanced bioavailability during neonatal development. BPA is present in the mouse fetus and amniotic fluid during maternal exposure in higher concentrations than that of maternal blood.
The U.S. EPA has established the safe daily intake of BPA to be 50 μg/kg body weight/d based on the assumption that the main source of exposure is oral through food ingestion. However, recent publications suggest that food is not the only relevant source of exposure and that the half-life of BPA in humans is longer than expected (6). Numerous publications addressing fetal exposures to BPA have used parenteral administration. This practice was based on one hand on the fact that the fetus is exposed to BPA through the internal milieu of the mother, and on the other hand that parenteral administration via an osmotic minipump allows for a precise and constant level of exposure. Using this route of administration, exposure of a pregnant mouse dam to 25 and 250 ng BPA/kg body weight/d (namely, 2000 and 200 times lower than the safe dose) for 14 d beginning on d 8 gestation has been shown to impact certain aspects of development in their female offspring. When examined on gestational d 18, fetuses of mothers exposed to the higher dose of BPA exhibited altered growth parameters of the mammary gland anlagen. Changes in the appearance of the mammary epithelium were observed, such as decreased cell size and delayed lumen formation, as well as increased ductal area. In the stroma, BPA exposure promoted advanced maturation of the fat pad and altered localization of fibrous collagen (128). Because maturation of the fat pad is the driving event for ductal growth and branching, it is likely that the increased ductal area in BPA-exposed animals is due to the accelerated formation of their fat pads. By postnatal d 10, in the offspring born to mothers exposed to either dose of BPA, the percentage of proliferating epithelial cells was significantly decreased relative to those not exposed. At 30 d of age, the area and number of terminal end buds relative to the gland ductal area increased, whereas cell death in these structures decreased in BPA-exposed offspring compared with controls. It is likely that the reduced cell death in the terminal end buds of BPA-exposed females may be the cause of the observed ductal growth delay because cell death is essential for both the hollowing and the outward growth of the subtending duct. Collectively, these effects observed at puberty may be attributed to an increased sensitivity to estradiol that has been observed in the BPA-exposed animals (145). Because of the new epidemiological data cited above and the effects found in the low-dose animal studies using parenteral exposure, the EPA recommendations need to be reevaluated.
In animals exposed perinatally to BPA, there was also a significant increase of ductal epithelial cells that were positive for progesterone receptor at puberty. These positive cells were localized in clusters, suggesting future branching points. Indeed, lateral branching was significantly enhanced at 4 months of age in offspring born to mothers exposed to 25 ng BPA/kg body weight/d (145). These results are compatible with the notion that increased sensitivity to estrogens drives the induction of progesterone receptors in epithelial cells, leading to an increase in lateral branching. By 6 months of age, perinatally exposed virgin mice exhibit mammary glands that resemble those of a pregnant mouse, as reflected by a significant increase in the percentage of ducts, terminal ends, terminal ducts, and alveolar buds (146). Additionally, intraductal hyperplasias, which are considered preneoplastic lesions, were observed starting at 3 months of age (147).
To explore the links between prenatal BPA exposure and mammary gland neoplasia, a rat model was chosen because it closely resembles the human disease regarding estrogen dependency and histopathology. BPA was administered to pregnant dams at doses of 2.5, 25, 250, and 1000 μg/kg body weight/d. Fetal exposure to BPA, from gestational d 9 to postnatal d 1, resulted in the development of carcinomas in situ in the mammary glands of 33% of the rats exposed to 250 μg/kg body weight/d, whereas none of the unexposed animals developed neoplasias (148). These cancers were only observed after the animals had reached young adult age. Fetal exposure to BPA significantly increased the number of precancerous lesions (intraductal proliferation) by three to four times, an effect also observed in puberty and during adult life. The lesions observed in the BPA-exposed animals were highly proliferative and contained abundant ER-positive cells, suggesting that the proliferative activity in these lesions may be estrogen mediated. Comparable preneoplastic lesions were found in a study using a different rat strain (149). Additionally, this study found stromal alterations such as desmoplasia and mast cell invasion; these features are often observed during neoplastic development. Moreover, when challenged with a subcarcinogenic dose of nitrosomethylurea, only the BPA-exposed animals developed palpable tumors (carcinomas). The period of vulnerability of the mammary gland to BPA does not cease at the neonatal stage. BPA exposure during lactation followed to exposure to the carcinogen DMBA resulted in mammary tumor multiplicity and reduced tumor latency compared with control animals (exposed solely to DMBA) (150). These results indicate that perinatal exposure to environmentally relevant doses of BPA results in persistent alterations in mammary gland morphogenesis, development of precancerous lesions, and carcinoma in situ. Moreover, the altered growth parameters noted in the developing mammary gland on embryonic d 18 suggest that the fetal gland is a direct target of BPA, and that these alterations cause the mammary gland phenotypes observed in perinatally exposed mice at puberty and adulthood.
In summary, exposure to estrogens throughout a woman’s life, including the period of intrauterine development, is a risk factor for the development of breast cancer. The increased incidence of breast cancer noted during the last 50 yr may have been caused, in part, by exposure of women to estrogen-mimicking chemicals that have been released into the environment from industrial and commercial sources. Epidemiological studies suggest that exposure to xenoestrogens such as DES during fetal development, to DDT around puberty, and to a mixture of xenoestrogens around menopause increases this risk. Animal studies show that exposure in utero to the xenoestrogen BPA increases this risk. Moreover, these animal studies suggest that estrogens act as morphogens and that excessive perinatal exposure results in structural and functional alterations that are further exacerbated by exposure to ovarian steroids at puberty and beyond. These altered structures include preneoplastic lesions, such as intraductal hyperplasias, and carcinomas in situ. Additionally, these mammary glands are more vulnerable than their normal counterparts to carcinogenic stimuli. Exposures to other endocrine disruptors that are not estrogenic, such as dioxins, were reported to increase breast cancer incidence in humans and to alter mammary gland development in animal models. Collectively, these data support the notion that endocrine disruptors alter mammary gland morphogenesis and that the resulting dysgenic gland becomes more prone to neoplastic development.
V. Male Reproductive and Developmental Health: The Human Evidence
A. Introduction to male reproductive health
The mechanisms through which environmental chemicals alter the endocrine system are elucidated through experimental animal studies and in vitro systems. In epidemiological studies it is generally not possible to explore potential mechanisms. Nevertheless, epidemiological studies are essential to our understanding of the potential risks, or lack thereof, of EDCs on human reproductive function and development.
Human evidence of altered male reproductive and developmental health in relation to EDCs is limited (Table 2 2). ). As has been shown in the recent Third National Report by the Center for Disease Control (151), humans are exposed, at a minimum, to hundreds of environmental chemicals, of which dozens are known EDCs. A major limitation of epidemiological studies is that they generally only measure human exposure to a single EDC, or at best to a set of isomers or congeners within a family of EDCs. A fuller understanding of potential human health risks requires studying the complex mixtures to which we are exposed. This limitation, already raised in other sections, should be kept at the forefront as the current epidemiological evidence on health risks from EDCs is presented.
For the purposes of this report, the male reproductive health endpoints under consideration include, among others: 1) disrupted reproductive function, manifest as reduced semen quality and infertility; 2) altered fetal development, manifest as urogenital tract abnormalities, including hypospadias and cryptorchidism; and 3) testicular germ cell cancer (TGCC).
B. Male reproductive function and development
1. TDS: A unifying hypothesis
Skakkebaek et al. (21) hypothesized that diminished semen quality, TGCC, and male urogenital tract anomalies may share a common causal pathway. They defined this triad as the TDS. The hypothesis invokes a common pathway by which EDCs, and other environmental chemicals and genetic factors, may lead to abnormal development of the fetal testis, producing testicular dysgenesis that can manifest as an increased risk of urogenital abnormalities in newborn males, as well as altered semen quality and TGCC in young men. As a cautionary note, the manifestations (or symptoms) of TDS have other causes apart from testicular dysgenesis.
It is hypothesized that TDS is due to prenatal Leydig and Sertoli cell dysfunction with secondary androgen insufficiency and impaired germ cell development. This should not be confused with the clinical diagnosis of dysgenetic testes, which is associated with genital ambiguity and a high risk of testicular malignancy (152). The existence of TDS as a distinct clinical entity and of possible associations with EDCs is an area of active research.
C. Semen quality: Temporal trends and EDC exposure
The epidemiological evidence on temporal trends in semen quality remains inconsistent. Some studies suggest that human semen quality has declined during the previous 50 yr (153,154,155), whereas other studies have not reported a decline (156,157,158). Despite the potential importance and relevance of early life exposure to EDCs, the epidemiological evidence on the relationship between semen quality and exposure to EDCs is limited to the assessment of adult exposure to EDCs. In the cases of PCBs, pesticides (persistent and nonpersistent), and phthalates, limited epidemiological evidence supports a relationship between adult exposure and reduced semen quality. However, most studies are cross-sectional in design; thus exposure and semen parameters were assessed at the same point in time. Although there are few studies in humans on the effects of developmental exposures to chemicals and semen quality in adulthood, this has been shown in animal models. Anway and Skinner (12) showed direct as well as transgenerational effects of EDCs on semen quality after intrauterine exposure.
1. Phthalates and semen quality
The diesters of 1,2-benzenedicarboxylic acid (phthalic acid), commonly known as phthalates, are a group of manmade chemicals widely used in industrial applications. They are primarily used as plasticizers in the manufacture of flexible vinyl plastic which, in turn, is used in consumer products, flooring, and wall coverings, food contact applications, and medical devices (159,160,161). They are also used in personal-care products (e.g., perfumes, lotions, cosmetics), as solvents and plasticizers for cellulose acetate, and in making lacquers, varnishes, and coatings, including those used to provide timed releases in some pharmaceuticals (159,162,163).
Human exposure to phthalates is widespread and occurs through ingestion, inhalation, and dermal contact (160,161,162,163,164,165). Parenteral exposure from medical devices and products containing phthalates are important sources of high exposure to phthalates, primarily di-(2-ethylhexyl) phthalate (DEHP) (161,166). Phthalates have biological half-lives measured in hours, are rapidly metabolized, and are excreted in urine and feces (160,161,162,163). The most common biomonitoring approach for investigating human exposure to phthalates is the measurement of urinary concentrations of phthalate metabolites.
There are few epidemiological studies on phthalates and semen quality. A large study on male partners of subfertile couples from an infertility clinic in Massachusetts (167,168) found associations between monobutyl phthalate (MBP; the hydrolytic metabolite of dibutyl phthalate) and below World Health Organization (WHO) reference value sperm motility and sperm concentration. There was also a dose-response relationship between monobenzyl phthalate (MBzP, the primary hydrolytic metabolite of butylbenzylphthalate) and below WHO reference value sperm concentration. In contrast to the U.S. study, in a Swedish study there were no relationships of MBP or MBzP with any of the semen parameters (169). Potential reasons explaining why the two studies found differing results include differences in age and fertility of the study populations. The Swedish study population consisted of young men (median age, 18 yr; range, 18–21 yr) from the general population, whereas in the U.S. study the median age of the men from an infertility clinic was 35.5 yr and ranged from 22 to 54 yr. None of the men from the infertility clinic were 21 yr of age or younger. Men presenting to an infertility clinic may be more “susceptible” to reproductive toxicants, including phthalates, than men from the general population. Furthermore, it is also unclear whether middle-aged men, compared with young men, are more susceptible to reproductive toxicants because of an age-related response to the toxicant.
2. PCBs and semen quality
PCBs are a class of synthetic, persistent, lipophilic, halogenated aromatic compounds that were widely used in industrial and consumer products for decades before their production was banned in the late 1970s. PCBs were used in cutting oils, lubricants, and as electrical insulators. As a result of their extensive use and persistence, PCBs remain ubiquitous environmental contaminants. They are biologically concentrated and stored in human adipose tissue. The general population is exposed primarily through ingestion of contaminated foods (e.g., fish, meat, and dairy products), because PCBs can bioaccumulate up the food chain. As a result of their persistence and ubiquity, measurable levels of serum PCBs are found in the majority of the U.S. general population (170). Serum levels of PCBs are an integrated measure of internal dose, reflecting exposure from all sources over the previous years; depending on the congener, the half-life of PCBs in the blood ranges from 1 to 10 or more years (171,172). Notably, there are 209 different possible chlorine substitutions on the biphenyl backbone of PCBs, with the resulting PCB molecules having different structural, functional, and toxicological properties (173,174).
The epidemiological evidence on the relationship between PCBs and semen quality support an inverse association of PCBs with reduced semen quality, specifically reduced sperm motility. Such relationships have been consistently reported across studies performed in different countries (India, The Netherlands, Taiwan, Sweden, and the United States). The associations were found across a range of PCB levels, suggesting that there was not a threshold. The PCB levels in these studies ranged from low background levels (175,176,177), to high background levels due to consumption of contaminated fish (178), to even higher exposure levels due to ingestion of contaminated rice oil (179,180).
3. Dioxins and semen quality
A recently published study of dioxin exposure and semen quality suggested that timing of exposure may have an impact upon the response (181). A chemical plant explosion in 1976 in Seveso, Italy, led to environmental contamination with high levels of TCDD. Exposed men in three age groups (1–9, 10–17, and 18–26 yr of age in 1976) were studied in 1998. Interestingly, the men exposed prepubertally (1 to 9 yr) had an inverse association between serum TCDD concentrations and semen quality, specifically sperm count and motility, whereas the men exposed at ages 10–17 yr had a positive association with semen quality, referred to as stimulatory by the authors. The men exposed at 18–26 yr of age had no associations of TCDD with semen quality. Men exposed at both 1–9 and 10–17 yr of age had lower estradiol and higher FSH concentrations compared with unexposed men. These results suggest that the timing of exposure, i.e., life stage, may have importance in determining the impact of environmental exposures.
4. Nonpersistent pesticides and semen quality
Nonpersistent pesticides (also referred to as “contemporary-use pesticides”) are chemical mixtures that are currently available for application to control insects (insecticides), weeds (herbicides), fungi (fungicides) or other pests (e.g., rodenticides), as opposed to pesticides that have been banned from use in most countries (e.g., many of the formerly popular organochlorine pesticides such as DDT). Three common classes of nonpersistent pesticides in use today include organophosphates, carbamates, and pyrethroids. Although environmentally nonpersistent, the extensive use of pest control in these various settings results in a majority of the general population being exposed to some of the more widely used pesticides at low levels. Exposure among the general population occurs primarily through the ingestion of foods that contain low levels of pesticide residue or through inhalation and/or dermal exposure in or around the home and in other indoor environments.
Several epidemiological studies suggest an association between nonpersistent pesticide exposure and altered semen quality. Most of the data are from occupational studies involving simultaneous exposure to several pesticides (182,183,184,185,186,187,188,189,190,191). Two recent studies found associations between pesticide exposures representative of the general population and reduced semen quality (192,193).
In a small study on male partners of pregnant women, Swan et al. (192) compared urinary concentrations of pesticide biomarkers in 34 men with sperm concentration, motility, and morphology below the median (defined as cases) to 52 men with above-median semen parameters (defined as controls). They found elevated odds ratios for poorer semen quality in relation to urinary concentrations of alachlor mercapturate, 2-isopropoxy-4-methyl-pyrimidinol (diazinon metabolite), atrazine mercapturate, 1-naphthol (carbaryl and naphthalene metabolite), and 3,5,6-trichloro-2-pyridinol (chlorpyrifos metabolite).
In a study among 272 men from an infertility clinic, Meeker et al. (193) found inverse associations between urinary levels of 1-naphthol, a metabolite of both carbaryl and naphthalene, with sperm concentration and motility. They also found a suggestive inverse relationship between the urinary metabolite of chlorpyrifos (3,5,6-trichloro-2-pyridinol) and sperm motility.
In summary, in addition to evidence from occupational studies, there are limited human studies suggesting reduced semen quality in relation to nonoccupational exposure to nonpersistent pesticides, specifically some herbicides and insecticides.
D. Male urogenital tract malformations
Epidemiological studies provide inconclusive evidence on temporal trends in cryptorchidism and hypospadias. Studies show that the prevalence of cryptorchidism is variable and geographically specific (194), with temporal upward trends noted in some studies but not others (15,195,196). The prevalence data for cryptorchidism are difficult to interpret because of the limitations of registry-based data and how they are obtained, changes in clinical practice that emphasize earlier diagnosis and treatment, confounding factors such as birth weight and prematurity, and inaccurate diagnosis related to changes in testicular position (spontaneous descent or secondary “ascent”) over time (197). Similarly, data for hypospadias prevalence are difficult to interpret. Although prevalence temporally increased in some locations, other reports showed no trends over time (195,198,199,200). Ascertainment bias may also easily exist for this anomaly, particularly for milder forms, because both false-negative and false-positive diagnoses may be made in newborns based on circumcision status.
Epidemiological evidence for EDC exposure and cryptorchidism or hypospadias is limited. Maternal serum concentrations of PCBs, DDT, or DDE (primary metabolite of DDT) were weakly associated or not associated with cryptorchidism or hypospadias in offspring (201,202,203,204).
The relationship of parental or general community pesticide exposure with hypospadias or cryptorchidism is suggestive (205,206,207,208,209,210), but there is the need for further research that explores maternal and/or paternal exposure to specific pesticides with urogenital anomalies.
In one of the only human studies on phthalates and male genital development, Swan et al. (23) determined “anogenital index” (anogenital distance/body weight) and testicular position in young boys (mean age, 16 months) and corresponding maternal levels of urinary phthalate metabolites at three separate clinical sites. In this study, the authors found significant inverse relationships between the highest maternal levels of MBP, MBzP, monoethyl phthalate, and monoisobutyl phthalate and anogenital index (odds ratio for MBP, 10.2; 95% confidence interval, 2.5–42.2), although MEP has not been linked to reproductive anomalies in rodent studies based on oral administration rather than transdermal, which is the route for human exposure via its use in personal-care products (197). The implication of a reduced anogenital index in rats is well defined, but the clinical implications of reduced anogenital index in human male infants is unknown.
In summary, the strongest epidemiological data that link EDC exposure to cryptorchidism and/or hypospadias are those suggesting an association between residency in agricultural areas and/or measures of direct parental exposure to nonorganochlorine pesticides, without providing insight into specific potentially causative agents. However, these data are not necessarily consistent for both anomalies or congruent with observations made in animal experiments. Further studies will be needed to provide a clearer understanding of the role(s) of specific EDCs in the etiology of genital anomalies in man.
E. Testicular germ cell cancer
Epidemiological studies show both geographical variability and dramatic recent upward trends in the incidence rate of TGCC (212,213,214,215,216). The steep temporal rise over a relatively short period of several decades suggests that genetic factors alone cannot explain it. Therefore, environmental and lifestyle factors have been hypothesized to play a role. Evidence for environmental and lifestyle factors is supported by migration studies in which the first generation of immigrants have incidence rates similar to their country of origin (birth), but their offspring had rates similar to men in the country in which they were born and raised (217).
The earliest suggestion of epidemiological evidence related to prenatal estrogen exposure and increased risk of TGCC came from a study in 1979 (218). However, other studies have not consistently confirmed these earlier results (219). At present, the evidence on EDCs and risk of TGCC is very limited. Interestingly, in a novel case-control study on EDCs and TGCC, Hardell et al. (220,221) did not find associations between serum concentrations of organochlorines among cases and controls and risk of TGCC, but instead found that blood organochlorine levels measured in their mothers, decades after their sons’ birth, were predictive of increased risk. The organochlorines measured included PCBs, p,p′-DDE (primary long-lived metabolites of DDT), and hexachlorobenzene, a fungicide. The study was small (44 case mothers and 45 control mothers), and the median time from the fetal period until blood sampling for the cases and controls was approximately 30 yr. It is important to keep in mind that despite the long period between the etiological relevant exposure window and measurement of organochlorines, their long half-lives, on the order of years to a decade, makes it possible to estimate historic exposure using the mothers’ blood samples. Therefore, the limited studies suggest that in utero exposure to environment EDCs represents the relevant etiological window of exposure. If this is borne out to be true, it will mean that epidemiologists need to consider innovative study designs to better assess prenatal exposure windows for endpoints that may not manifest for decades. Prospective pregnancy cohort follow-up studies for TGCC would be difficult and costly to implement because TGCC is a rare cancer and prospective study would require unrealistically large cohorts.
F. Conclusions
This section has tried to provide highlights and insights into the current state of the epidemiological evidence on the relationship between EDCs and male reproductive and developmental health. The overview was not meant to be an exhaustive review of the evidence, but rather a synthesis of the current knowledge in an ever-changing field of inquiry and discovery. Although there is current scientific, public, and governmental interest in the potential health risks of exposure to EDCs, the human evidence on associations of EDCs with altered male reproductive health endpoints remains limited and, in certain instances, inconsistent across studies. This highlights the need for further epidemiological research on these classes of EDCs.
VI. Prostate Cancer
A. Introduction to prostate cancer
Prostate cancer is the most common solid cancer in males and the second leading cause of cancer deaths in American men (222). In addition, benign prostatic hyperplasia is the most common benign neoplasm, occurring in approximately 50% of all men by the age of 60. The basis for these high rates of abnormal prostatic growth is not well understood despite decades of extensive research on the topic. Nonetheless, it is accepted that steroids play a fundamental role in the initiation and progression of prostate cancer, which forms the basis for hormonal treatment strategies. Men who have undergone early castration do not develop prostatic carcinoma (223). Charles Huggins received the Nobel Prize for his work revealing that regression of prostate cancer can be initially achieved by castration and androgen blockade (224). In addition to androgens, it has been proposed that estrogens are involved in the etiology of benign prostatic hyperplasia and prostatic cancer (225,226,227), and the use of antiestrogens has been recently recognized to have a therapeutic role in prostate cancer management (228,229). The prostate gland contains both ERα and ERβ during development and into adulthood, with ERα primarily found in stromal cells and ERβ in differentiated epithelium (230). It is also believed that prostatic developmental events under the regulation by steroids early in life may be linked to the predisposition of this structure to high rates of disease in adult men (231,232). Moreover, the prostate gland is particularly sensitive to estrogen exposures during the critical developmental period relative to adult estrogenic responses (233).
The established risk factors for prostate cancer are age and race. African-American men have the highest incidence of prostate cancer worldwide, at rates 2-fold those for Caucasian-American counterparts. Family history (genetics), diet, and environmental factors are also recognized to impact prostate cancer risk. However, in the human population, direct connections between EDCs and prostate cancer risk have not been established. Due to the hormonal basis of this disease and the evidence that dietary compounds high in phytoestrogens (e.g., genistein) can control prostate cancer growth in humans, there is reasonable cause to evaluate and understand any potential relationship between environmental EDCs and prostate cancer risk. Because there are difficulties in directly associating prostate cancer risk in humans with EDC exposures, potential risk(s) will have to be ascertained from research with animal models, particularly those that are responsive to environmentally relevant exposures. The sections below summarize the evidence obtained from epidemiological studies, in vitro studies with human prostate cells, and in vivo studies in animal models that indicate associations between EDCs and prostate cancer, carcinogenesis, and/or susceptibility (Fig. 1 1 ).
B. Evidence and mechanisms for EDC effects on the prostate
1. Farming and pesticides
The most compelling data for a link between prostate cancer and environmental factors outside of diet in humans comes from the established occupational hazard of farming and increased prostate cancer rates (234,235,236). Although several variables may contribute to this association, chronic or intermittent exposures to pesticides are the most likely explanation (236,237). A large epidemiology study (Agricultural Health Study) conducted collaboratively between the National Cancer Institute, the National Institute of Environmental Health Sciences, and the EPA examined agricultural lifestyles and health in approximately 90,000 participants in North Carolina and Iowa since 1993 (www.aghealth.org). Evaluation of more than 55,000 pesticide applicators revealed a direct link between increased prostate cancer rates and exposure to methyl bromide, a fungicide with unknown mechanism of action (236). In addition, six pesticides (of 45 common agricultural pesticides) showed significant correlation with exposure and increased prostate cancer rates in men with a familial history of the disease, suggesting gene-environment interactions. These six agents were chlorpyrifos, fonofos, coumaphos, phorate, permethrin, and butylate (236,238). The first four compounds are thiophosphates that share a common chemical structure. These agents are acetylcholine esterase inhibitors and have not been shown to have direct estrogenic or antiandrogenic activities. However, a literature search found that these compounds have marked capacity to inhibit p450 enzymes. Chlorpyrifos, fonofos, and phorate strongly inhibit CYP1A2 and CYP3A4, which are the major p450s that metabolize estradiol, estrone, and testosterone in the liver (239,240). Thus it is possible that exposure to these compounds can interfere with metabolism of steroid hormones and, in so doing, disturb the normal hormonal balance that might contribute to increased prostate cancer risk. A similar mechanism of endocrine disruption in vivo has been identified for PCBs and polyhalogenated aromatic hydrocarbons (including dioxins, BPA, and dibenzofurans) through marked inhibition of estrogen sulfotransferase, which in turn elevates bioavailable estrogens in target organs (45,241).
2. Environmental estrogens
In men, chronically elevated estrogens have been associated with increased risk of prostate cancer (227). In rodents, natural estrogens combined with androgens induce prostate cancer (225,242). For simplicity, we herein refer to environmental estrogens as molecules with identified estrogenic activity (estrogen mimics), primarily through ER activation.
a. DES.
In utero DES exposure is an important model of endocrine disruption and provides proof-of-principle for exogenous estrogenic agents altering the function and pathology of various end-organs. Maternal usage of DES during pregnancy resulted in more extensive prostatic squamous metaplasia in human male offspring than observed with maternal estradiol alone (243). Although this prostatic metaplasia eventually resolved during postnatal life, ectasia and persistent distortion of ductal architecture remained (244). These findings have led to the postulation that men exposed in utero to DES may be at increased risk for prostatic disease later in life (245), although the limited population studies conducted to date have not identified an association (245). Nonetheless, several studies with DES in mouse and rat models have demonstrated significant abnormalities in the adult prostate, including increased susceptibility to adult-onset carcinogenesis after early DES exposures (246,247,248,249). It is important to note that developmental exposure to DES, as with other environmental estrogens, has been shown to exhibit a biphasic dose- response curve with regard to several end-organ responses, and this has been shown to be true for prostatic responses as well (250). Low-dose fetal exposure to DES or BPA (see below) resulted in larger prostate size in adulthood compared with controls, an effect associated with increased levels of prostatic ARs. This contrasts with smaller prostate sizes, dysplasia, and aging- associated increases in carcinogenesis found after perinatal high-dose DES exposures as noted above. This differential prostatic response to low vs. high doses of DES and other EDCs must be kept in mind when evaluating human exposures to EDCs because the lack of a response at high doses may not translate into a lack of negative effects at low, environmentally relevant doses of EDCs.
b. BPA.
BPA is a synthetic monomer used in the production of polycarbonate plastics and epoxy resins and is one of the highest production synthetic compounds worldwide. Importantly, conjugated BPA was detected in the urine of 93% of the U.S. population in a recent screen conducted by the Center for Disease Control. Although the relative binding affinity of BPA for ERα and ERβ and its capacity to activate ER-dependent transcription is approximately 1,000 to 10,000 lower than estradiol or DES (1,251), BPA was capable of activating an estrogen-responsive luciferase reporter at levels that were 50% of 17β-estradiol activation (252). Thus, whereas BPA may have a significantly lower potency than endogenous estrogens in vitro, it is a full agonist for both ERα and ERβ. Furthermore, BPA induces ER through nongenomic pathways with an EC50 equivalent to 17β-estradiol, suggesting that in vivo estrogenic activity of BPA may be due to nongenomic activation of ER (253,254).
The carcinogenic potential of BPA was recently evaluated by an expert panel convened by the EPA and the National Institute of Environmental Health Sciences, and the written report, which includes prostate cancer findings, has been published (255). In summary, there is evidence using in vitro prostate cell cultures and rodent models showing that BPA can modulate prostate cell proliferation and increase susceptibility of the prostate gland to hormonal carcinogenesis. Using transcriptional assays, BPA (1 n m ) was found to activate a mutated AR (AR-T877A) that is frequently found in advanced prostate cancers of patients who relapsed after androgen deprivation therapy (256). Furthermore, BPA exposure led to unscheduled cell cycle progression and cellular proliferation in the absence of androgen in LNCaP cells that expressed this mutant AR. Because BPA had no impact on wild-type AR, these findings demonstrate that the common gain-of-function AR mutant had attained the ability to utilize BPA as an agonist. Importantly, the BPA effects were greatest at lower doses of BPA compared with high-dose exposures. In vivo analyses of the impact of BPA on human prostate tumor growth and recurrence was performed utilizing a xenograft model (257). At low doses equivalent to human exposures, prostate tumor size increased after BPA exposure when compared with placebo control mice. Additionally, mice in the BPA cohort demonstrated an earlier rise in prostate-specific antigen (biochemical failure), which indicates that BPA significantly shortened the time to therapeutic relapse. These outcomes underscore the need for further study of the effects of BPA on tumor progression and therapeutic efficacy.
Recent studies using a rat model have shown that early-life exposure to environmentally relevant levels of BPA can increase susceptibility to prostate carcinogenesis, possibly by developmentally reprogramming carcinogenic risk (122,258). Rats were exposed to low doses of BPA (10 μg/kg body weight) during the early postnatal period when the prostate undergoes morphogenesis. In adulthood, estradiol levels were elevated 3-fold through the use of implants for 16 wk. Rats exposed neonatally showed a significant increase in the incidence (100 vs. 40%) and grade of prostatic intraepithelial neoplasia lesions compared with rats neonatally exposed to oil alone. These lesions in BPA-exposed rats exhibited high levels of proliferation and apoptosis suggestive of perturbed homeostasis leading to pathological lesions. Furthermore, prostates from BPA-exposed animals were shown to have permanent epigenetic changes with altered DNA methylation patterns in multiple genes that resulted in altered gene transcription. Together, these findings indicate that BPA may “imprint” the prostate through epigenetic modifications, resulting in predisposition to carcinogenesis.
c. PCBs.
PCBs are persistent organic pollutants that are fat-soluble and bioaccumulate in human body fat deposits. Many PCBs have estrogenic or antiandrogenic activity and as such, may perturb the prostate gland. A recent analysis in Swedish men with and without prostate cancer of adipose tissue PCB concentrations revealed a significant association between PCB levels in the higher quadrants and prostate cancer odds ratio, with the most marked associations for PCB 153 and transchlordane (259). An extensive epidemiological study of capacitor manufacturing plant workers exposed to high levels of PCBs revealed a strong exposure-response relationship for prostate cancer mortality (260). These results support previous findings of correlations between PCB 153 and 180 and prostate cancer risk in electric utility workers (261,262). Although estrogenic activity of these compounds is a suspected mode of action, there is also evidence that PCBs inhibit estrogen sulfotransferase activity in the liver and effectively increase bioavailable estrogen in the body (45). Further investigation using animal models is warranted for PCBs and prostate cancer risk.
d. UV filters.
Recent reports have shown that UV light filters used to protect against the sun have estrogenic activity (263). In particular, 4-methylbenzylidene camphor and 3-benzidene camphor are ERβ ligands (264). Although there are no studies on these UV filters and human prostate cancer, two reports indicate that early life exposure to these compounds can alter prostate gland development, growth, and gene expression in the rat prostate (263,265). Thus, it is possible that the fetal prostate in humans may be affected after maternal use of these compounds, although this remains to be examined.
e. Cadmium.
Cadmium has been shown to act as a ligand for the ER and function as an estrogenic mimic. Although some large epidemiological studies indicated a relationship between cadmium exposure and rates of prostate cancer, these findings have been challenged in other reports (266). Cadmium has been shown to have proliferative action on human prostate cells in vitro through an ER-dependent mechanism, and this exposure was associated with progression to androgen independence (267). In addition, prostatic tumors have been experimentally induced by oral exposure to cadmium (268). Because cadmium bioaccumulates in the body, further epidemiological analysis of cadmium and prostate cancer risk is warranted, particularly in men with occupational exposures.
f. Arsenic.
Exposure to arsenic has long been associated with a number of diseases, including cancers (269). More recently, it has been documented that arsenic may mediate some of these effects through endocrine disruption, specifically through interaction with ERs and activation of estrogen-regulated genes (270). A recent report has found that arsenic induced malignant transformation of prostate epithelial cells in vitro, driving them toward an androgen-independent state (271). Progression to androgen-independent growth was shown to be mediated through Ras-MAPK pathways, and thus, it is possible that membrane ERs may mediate this effect. Epidemiological studies have shown an association between arsenic exposure and prostate cancer mortality in Taiwan (272), a finding that was substantiated in a more recent study in the United States (273). Thus it remains a possibility that endocrine disruption by arsenic can contribute to prostate cancer risk, and further research on this topic is essential.
3. Antiandrogens
Endocrine disruption that might affect the prostate gland can also be derived through antiandrogenic pathways. Because prostate cancer is an androgen-dependent disease, a brief review of known effects of some of these agents on the prostate gland is presented.
a. Vinclozolin.
Vinclozolin is a fungicide that is used as a pesticide on crops. It possesses known antiandrogenic properties through interference with AR activity (274). Rats exposed to vinclozolin during early development were reported to have reduced prostate gland growth and size (275). Recently, maternal exposure to vinclozolin was shown to produce transgenerational effects with adverse consequences on the prostate gland, including atrophy and prostatitis for four generations (34,276). However, because vinclozolin functions through AR antagonism, it is unexpected that vinclozolin will lead to prostate cancer.
b. DDT/DDE.
DDT and its metabolic derivative p,p′-DDE were widely used as pesticides in the United States, and their use is still in effect in other countries. Although many reproductive abnormalities have been found with DDT/DDE, there is no known association between its exposure and prostate cancer risk (277). Again, due to its antiandrogenic actions, it is not expected to drive prostate cancer. A number of key questions remain unresolved but merit future investigation, not just in prostate cancer but in other fields (Boxes 1 and 2). Spanning from the molecular to the clinical, they highlight the need for a better understanding of the pathogenesis of prostate cancer and the potential role of EDCs in this process.
BOX 1. Recommendations for research on prostate cancer
It remains unclear whether EDC exposures directly induce or promote prostate cancer. If either occurs, it will be necessary to determine the mode of action.
It will be important to determine whether estrogenic or antiandrogenic EDCs modulate disease risk or progression in the adult male. One possibility may be that EDC exposure may influence prostate cancer susceptibility in subpopulations of men. If so, it would be important to determine the other risk factors that EDCs might synergize with to influence prostate cancer incidence and/or progression.
It is unknown whether there is an additive or synergistic effect from EDC mixtures and prostate cancer risk or growth.
It is necessary to determine whether the in utero developing human prostate is sensitive to EDCs and whether this may influence the prostate cancer risk in the aging male.
Epidemiology studies need to be undertaken to evaluate the long-term outcome for prostate cancer incidence, grade, stage, and progression in DES-exposed sons.
An unexplored and important issue is whether there may be a transgenerational risk for prostate cancer as a function of EDC exposures.
Are there epigenetic pathways that mediate developmental exposures to EDCs and prostate disease with aging?
It will be important to establish molecular markers for EDC exposures as they relate to prostate disease risk.
BOX 2. Recommendations for research and practice regarding EDCs
In newborns with IUGR and/or anomalies of sexual differentiation including cryptorchidism and hypospadias, screen for EDCs in maternal serum and in breast milk, and archive biological samples for further screening.
Prioritize a search for early (i.e., neonatal period and infancy) biomarkers of EDC effects and early indicators of exposure to EDCs during fetal life.
Identify groups at high or low risk of exposure to EDCs for prospective studies correlating indicators of early exposure with subsequent clinical characteristics throughout infancy, childhood, and adolescence.
Search for and study polymorphisms in enzymes (e.g., CYP enzymes) that predispose groups/individuals to greater/lesser vulnerability to EDCs.
Identify the chemical or chemicals in pesticides that negatively impact risk for prostate cancer, breast cancer, endometriosis, and others in humans.
Molecular studies in vitro and with in vivo animal models are needed to identify pathways for EDC influence on endocrine tissues.
The mechanisms by which EDCs affect neuroendocrine systems need to be ascertained. In addition, studies on EDCs on several of these systems are very underrepresented, and these fields need to be expanded.
The transgenerational, epigenetic effects of EDCs need to be much more broadly studied across different endocrine and reproductive systems.
The interaction of EDCs with central nervous system developmental processes dependent on thyroid hormones (e.g., cochlear development) or sex steroids (e.g., hippocampal development) warrant early in vitroand in vivostudies.
The effects in animal stem cells or progenitor cells in different tissues could decipher EDC-sensitive genes possibly used as reporters in early biomarking.
There is a gap in knowledge about the mechanisms by which EDCs act as ″obesogens,″ particularly in how these processes develop.
Large prospective epidemiological studies need to be undertaken to examine the relationships between EDC exposures, particularly agents with estrogenic and antiandrogenic activity, and relevant endpoints as identified in this report. The National Children’s Study will be especially critical to this undertaking.
Identify populations or subgroups with high exposures to EDCs and conduct exposure-response studies among these populations.
Perform epidemiological studies that incorporate measurement of exposure to multiple EDCs, allowing for the study of human health effects from chemical mixtures.
Develop and incorporate validated biomarkers of EDC exposures and relevant outcomes into new and ongoing epidemiological studies.
Observations from occupational and environmental exposures in humans and their corresponding disease states should inform what animal studies should be performed and which EDCs should be studied, and should be used to inform policy decisions regarding human exposures to EDCs.
Set up early detection programs for testis cancer in the follow-up management of infertile men with poor semen quality.
Take a careful history of onset of reproductive disorders along with an occupational and environmental exposure history.
Think ″epidemiologically″ about the patients: that is, consider possible exposure to EDCs in geographical or community subgroups showing unexpectedly high prevalence of any of the disorders possibly related to EDCs.
Clinicians can advise patients about exposures, minimizing risks, and abiding by the ″precautionary principle″ to preserve their reproductive health and that of generations thereafter.
Health care professionals need to be educated in sources and effects of environmental contaminant exposures in utero and across the life span.
Health care professionals need to have access to straightforward and accurate health information tools to share with patients.
Clinicians should be made aware of the potential risks posed by EDCs. This would, for instance, help them to seek evidence for exposure when treating patients presenting with early thelarche or puberty.
VII. Neuroendocrine Targets of EDCs
The central neuroendocrine systems of the body serve as an interface between the brain and the endocrine systems in the rest of the body. These neuroendocrine systems control diverse functions such as reproduction, stress, growth, lactation, metabolism and energy balance (including thyroid), osmoregulation, and other processes involved in homeostasis. Considering that these neuroendocrine systems mediate the ability of the organism to respond to its environment through rapid (neuronal) and more sustained (endocrine) responses, it is not surprising that they are targeted by environmental EDCs (reviewed in Refs. 7, 278, and 279). Furthermore, neuroendocrine cells in the brain have both neuronal and endocrine properties, which is important in the context of endocrine disruption because EDCs can have neurobiological and neurotoxic effects (279), along with the endocrine effects discussed in this Scientific Statement.
The physiological processes controlled by central neuroendocrine systems are highly complex, making an understanding of neuroendocrine disruption a particular challenge. Each of these neuroendocrine systems comprises several interdependent levels of organization: the brain (specifically the hypothalamus), the pituitary gland, and often a target organ. These levels of organization may each produce a unique hormone(s) or a complex protein (e.g., breast milk), and each level also responds to the hormones produced by the other levels via feedback mechanisms (280). Here, we will discuss the evidence for central neuroendocrine systems as targets for EDCs (Fig. 1 1). ). The bulk of the literature to date has studied primarily the reproductive (HPG) system and secondarily the thyroid neuroendocrine system. The latter will be considered in detail in Section VIII, so the former (reproductive neuroendocrinology) will be the focus of the current discussion. Other neuroendocrine systems remain understudied and are only briefly mentioned. Nevertheless, they merit much more investigation in the future.
A. Endocrine disruption of reproductive neuroendocrine systems
1. GnRH neurons
Of the neuroendocrine systems, the reproductive HPG axis is best studied in the arena of endocrine disruption. The control of reproductive neuroendocrine function involves a group of neurons in the basal hypothalamus that synthesize and release the decapeptide GnRH (281). GnRH release drives reproduction throughout the life cycle, and this is the primary stimulus to the rest of the reproductive axis (the pituitary and gonads). GnRH release stimulates gonadotropin release from the anterior pituitary gland, which in turn activates steroidogenesis and gametogenesis in the ovary and testis. Steroid hormones produced by the gonad act on other target tissues that express estrogen, progestin, and/or ARs, a concept that is fundamental to endocrine disruption because so many EDCs act to interfere with steroid hormone actions. A second important concept is that sex steroids also control the hypothalamic GnRH neurons, but this involves indirect effects because GnRH neurons do not express most of the receptors for steroid hormones (282). This introduces the important point that other cells in the brain that express steroid hormone receptors and that regulate GnRH cells through afferent neural inputs are targets for EDCs. These points also relate to evidence that EDCs can act upon neurotransmitter systems that, at first glance, may not seem to have relevance to neuroendocrine control. For example, EDCs have been shown to cause neurotoxicity of noradrenergic, serotonergic, dopaminergic and other neurotransmitter-containing neurons (reviewed in Refs. 2 and 279). Considering that all of these neuronal types have been shown to express steroid hormone receptors and all of these cell types can project to and regulate GnRH neurons (281), this is a mechanism for convergence of effects of EDCs on the link between neural and endocrine systems.
One of the biggest challenges with the neuroendocrine system is gaining access to it. Hypothalamic neuroendocrine cells such as GnRH neurons are located in the hypothalamus at the base of the brain, making them difficult to access in animal models and impossible in humans. The hypothalamic-releasing hormones are not released in sufficiently high quantities to be detectable in peripheral circulation. Therefore, assays of hypothalamic function rely on hormone measurements of their corresponding pituitary hormones. If the pituitary sensitivity to hypothalamic output is compromised, then it is impossible to distinguish a primary hypothalamic or pituitary effect of an EDC. This has necessitated the use of animal models or in vitro assays to directly ascertain effects of EDCs on neuroendocrine peptide gene expression or release.
A reliable model for the GnRH system is the hypothalamic GT1 cell lines that have been used for nearly two decades as a proxy for the GnRH neuron in vivo (283). For example, PCBs (284) and organochlorine pesticides (methoxychlor, chlorpyrifos) (285) have been tested in this context. Application of these EDCs to GT1 cells caused significant changes in GnRH gene expression, GnRH peptide release, and the morphology of the GT1–7 cells. Interestingly, these substances often acted by nonlinear dose-response curves, with intermediate dosages exerting the greatest effects, typical of hormonally-active substances (286,287). Moreover, unlike traditional toxicological studies, effects of these environmental contaminants were in many cases stimulatory to the GnRH response. When comparisons were made to estradiol, at least some of the effects of these EDCs mimicked effects of estrogens on GT1 cell morphology, proliferation, and gene expression. In addition, blockade of ERs with ICI 182,780 diminished some of the actions of EDCs. Together, these data suggest that EDCs may directly target GnRH cell lines. Nevertheless, caution must be taken in interpreting these data, because the GT1 cells express some molecules not detectable in the animal’s GnRH cell, including some nuclear steroid hormone receptors. Other cell line models for neuroendocrine cells are available and may be useful in screening substances for neuroendocrine disrupting activities.
Studies using explanted hypothalamic dissections in a perifusion model from 15-d-old female rats tested effects of several EDCs on glutamate-evoked GnRH release, the latter model used as a reliable way of stimulating GnRH secretion (288). Of the EDCs tested, o,p′-DDT had the greatest stimulation of glutamate-evoked GnRH release, and BPA had a lesser effect. Not all EDCs were stimulatory; methoxychlor and p,p′-DDE had no effect in this in vitro model. Collectively, these data suggest EDC effects on GnRH release in a hypothalamic explant model (288). Finally, antagonists to the ER or AhR blocked effects of DDT, suggesting mediation of these endocrine-disrupting properties by these nuclear receptors and invoking a potential mechanism of action. In another study, this same group showed that DDT, but not DDE, decreased the interpulse interval of GnRH pulses (i.e., increased pulse frequency), again consistent with stimulatory effects of these EDCs on GnRH release (27).
Mammalian in vivo studies also implicate GnRH neurons as targets for EDCs. O’Byrne’s laboratory (289) has shown that coumestrol suppresses LH release (a proxy for GnRH) and the GnRH pulse generator. Bourguignon’s laboratory (27) reported that DDT accelerated the timing of puberty in female rats and altered the LH response to GnRH, although surprisingly, this was decreased in the DDT animals. Not all aspects of GnRH function are affected by all EDCs: Patisaul et al. (290) showed that expression of the immediate early gene fos in GnRH neurons was not altered by neonatal genistein or BPA. By contrast, data from Gore’s laboratory (291) suggest that EDCs can stimulate GnRH mRNA levels in laboratory rats. In the rabbit, prenatal vinclozolin (an endocrine-disrupting fungicide) decreased numbers of GnRH neurons in selected brain regions (292). Together, these results suggest actions of EDCs on GnRH neurons, although much more research is necessary to reconcile these data and better understand the mechanisms. These findings are not really surprising, considering that GnRH neurons act as the interface between endocrine and neural systems, but they are important because they show this level of regulation with the HPG axis.
Studies in fish also demonstrate effects of EDCs on the GnRH system. The strongest work has been published by the collaboration of Khan and Thomas (293). Using the Atlantic croaker as an experimental model, these labs showed that PCBs decreased preoptic-hypothalamic GnRH content, pituitary GnRH receptors, and the LH response to GnRH challenge (293). This effect was mimicked by an inhibitor of serotonin synthesis suggesting the possible mediation of effects of PCBs by the serotonergic pathway.
2. EDC effects on sexually dimorphic brain regions and behavior
The regions of the hypothalamus that control reproductive neuroendocrine systems undergo development during specific time periods, in large part due to exposures to endogenous steroid hormones such as estrogens and androgens. Although this is a simplification, it is speculated that the brains of male mammals become masculinized and defeminized due to actions of estradiol and testosterone produced by the developing (embryonic and early postnatal) testis. In female rodents, the best-studied model for endocrine disruption, the ovary is relatively quiescent during these developmental periods, and their brains are thought to be feminized and demasculinized due to the relative absence of exposure to these steroid hormones (reviewed in Ref. 294). However, it is important to note that the human ovary does produce estradiol (295), so there are species differences. Nevertheless, the developmental basis of adult disease applies to the development of the reproductive neuroendocrine system through actions during critical periods of sexual differentiation.
It should be apparent that exogenous hormones that may perturb steroidal actions through actions such as binding to steroid receptors, changing steroid metabolism, and others would have effects on the developing neuroendocrine system in a sexually dimorphic manner. There has been considerable and consistent research that shows that PCBs, phytoestrogens, fungicides, pesticides, and other xenobiotics can disrupt brain sexual differentiation (294). This type of disruption has a high likelihood of affecting both reproductive physiology and behavior later in life, and indeed, there is strong evidence in rodent models that reproductive success is diminished as a consequence (reviewed in Ref. 7). Early life exposure (late embryonic and/or early postnatal) to low doses of PCBs (296,297,298) or soy (299) significantly and adversely affected mating behaviors in female rats. Early postnatal treatment with coumestrol (a phytoestrogen) diminished masculine and feminine sexual behaviors (300,301). These results are consistent with a functional outcome for effects of EDCs in the neuroendocrine hypothalamus.
A recently published collaborative study demonstrated significant effects of prenatal vinclozolin on mate preference behavior in F3 descendents. In brief, pregnant rats were treated with vinclozolin or vehicle. The F1 vinclozolin male offspring developed latent disease in adulthood, consistent with the developmental basis of adult disease (35). Moreover, this phenotype was passed on to subsequent generations (through F3) via paternal germline transmission, due to epigenetic modification of specific genes. We evaluated the attractiveness of the male F3 vinclozolin descendents in comparison to F3 vehicle descendants in a mate preference test in which females were given the opportunity to spend time with a descendent of both treatments. The results showed a profound difference, with females spending significantly more time with an F3-vehicle compared with an F3-vinclozolin descendant male (302). These results show differences in behavior, as well as evolutionary impact on mating success, caused by endocrine disruption. Notably, these F3 descendants had no personal body burden or exposure to vinclozolin, and it has been postulated that the basis of the discrimination in the mate choice test was due to a transgenerational, epigenetically transmitted trait (302).
B. Hypothalamic-pituitary-adrenal (HPA) effects of EDCs
As articulated by Harvey et al. (303), “The adrenal is arguably the neglected organ in endocrine toxicology, and the lack of recognition of the importance of adrenal function in a regulatory endocrine disruption context and the need for an adrenal toxicology assessment strategy has been pointed out.” Numerous pharmaceuticals can affect the HPA axis, but this phenomenon has not, to our knowledge, been systematically studied for EDCs. Findings that the HPA axis is sensitive to HPG hormones suggest a potential mechanism by which EDCs may disrupt the HPA axis as well. Alternatively, EDCs may act directly upon the glucocorticoid or mineralocorticoid receptors or on steroidogenic pathways. EDCs including PCBs, dioxin, lindane, and others can affect synthesis of adrenal steroids, but specific effects on the neuroendocrine control of HPA function are lacking (303). This is an important area for future research.
C. Thyroid, metabolism, and growth
The hypothalamic-pituitary-thyroid (HPT) axis provides a critical test of the developmental basis of adult disease hypothesis because normal development and the acquisition of adult functions are dependent upon a euthyroid environment in the developing organism (304). Just a few examples are provided in this section because Section VIII provides a comprehensive review of endocrine disruption of thyroid systems. In rats, PCB congeners can affect the HPT axis at several levels, including a reduction in the T4 or TSH response to TRH (305). Low-dose exposure of pregnant rats to polybrominated diphenyl ether (PBDE) on d 6 of gestation reduced T4 levels in both dams and offspring (the latter measured on postnatal d 22), although it is unknown whether this is due to direct thyroid or neuroendocrine actions (306). Gray seals with higher blubber concentrations of industrial organochlorine compounds have lower total and free T3 concentrations (307). Considerably more information on the subject of thyroid disruption is provided in Section VIII.
The control of metabolism and energy balance extends well beyond the HPT axis. In the context of endocrine disruption, there are reports on effects of fetal DES, the prototypical estrogenic endocrine disruptor, on obesity in adulthood and even on a successive generation of mice (308). Although the exact mechanisms for such effects are not understood, the fact that the hypothalamus contains a complex neural circuitry that regulates energy and metabolic homeostasis suggests the possibility for this being a neuroendocrine action. Further discussion of this topic is in Sections VIII and IX of this Scientific Statement.
To our knowledge, there is little published work on neuroendocrine disruption of somatic growth. Although studies in fish show reductions in the gonadosomatic index, animals exposed to refuse or water waste (309), the mechanism for these effects and the respective roles of the growth, as opposed to the reproductive, axes are not known.
D. Hormonal targets of neuroendocrine disruption
There are both hormonally dependent and independent pathways by which EDCs exert neuroendocrine actions. EDCs may act upon nuclear hormone receptors that are expressed in hypothalamic or pituitary cells, thereby exerting feedback effects. Steroid hormone receptors are expressed abundantly in hypothalamus and other brain areas that control neuroendocrine functions (310,311,312). Along with “classical” nuclear steroid hormone-mediated actions, EDCs may exert actions via membrane steroid receptors (313,314) (reviewed in Ref. 315). These and other steroid-sensitive pathways are obvious targets by which EDCs act upon neuroendocrine systems.
The neuroendocrine actions of EDCs may occur via nonhormonally mediated mechanisms. Numerous neurotransmitter systems such as dopamine, norepinephrine, serotonin, glutamate, and others are sensitive to endocrine disruption (reviewed in Ref. 2). This point is important because it explains neurological effects of EDCs on cognition, learning, memory, and other nonreproductive behaviors, but it may also relate to reproductive neuroendocrine systems. As already mentioned, these neurotransmitters may coexpress steroid hormone receptors, so this steroid-sensitive circuitry may be an important target of EDC actions on neurotransmission.
Neuroendocrine systems are critically involved in the control of vertebrate homeostasis and physiology. Although we tend to think of them as independent systems, in fact there is considerable cross-talk among them. This is an important consideration in determining effects of EDCs; whereas no discernible effect may be determined in one system, it is important to evaluate the other systems for subtle but physiologically relevant effects. Therefore, there is a great need for additional interdisciplinary research on effects of EDCs in neuroendocrine systems. However, high-throughput assays for neuroendocrine effects of EDCs are difficult to develop due to the nature of these complex physiological systems. For example, it is impossible to test the “developmental basis of adult disease” hypothesis in a cell line. Animal studies are by nature labor intensive, particularly when they necessitate exposures during critical periods and when performed in species that give birth to litters as opposed to individuals, an intrauterine organization that is very different from the situation in humans. Thus, carefully designed neuroendocrine studies on EDCs in rodents need to take the litter composition and intra- and interlitter variability into consideration.
VIII. Thyroid Disruption
A. Introduction to thyroid systems
Thyroid hormone is essential for normal brain development, for the control of metabolism, and for many aspects of normal adult physiology. Therefore, changes in the function of the thyroid gland or interference with the ability of thyroid hormone to exert its action may produce effects on development, metabolism, or adult physiology. The goal of this section is to provide a brief overview of the literature regarding the mechanisms by which environmental chemicals may interfere with thyroid hormone action, which will require a brief background of thyroid endocrinology. In addition, we will describe some of the information in humans that indicate the extent to which environmental chemicals may be acting on thyroid hormone signaling in humans.
B. Environmental chemicals impacting thyroid function
A large number of industrial chemicals have been shown to reduce circulating levels of thyroid hormone. Brucker-Davis (316) and Howdeshell (317) have extensively reviewed this topic. Howdeshell categorized these chemicals (more than 150 in all) according to the mechanism by which the chemical was known to cause a reduction in serum thyroid hormone (see Table 1 1 in Ref. 317 for a full list). This point serves to illustrate clearly that there are many industrial chemicals that can interfere with thyroid function by acting on different points of regulation of thyroid hormone synthesis, release, transport through the blood, metabolism of thyroid hormone, and thyroid hormone clearance. In addition, many natural substances are known to affect thyroid function, including low iodine as well as goitrogens in various foods (318,319). The current section on thyroid disruption will emphasize the mechanisms by which chemicals are known to interfere with thyroid hormone action and highlight some recent information on the effects of chemicals on thyroid hormone receptors.
The first step in thyroid hormone synthesis is the uptake of iodide into the thyrocyte by the sodium/iodide symporter (NIS) (320). Iodine is essential for thyroid hormone synthesis, and iodine deficiency is an important public health problem worldwide (321). Thus, chemicals that interfere with the NIS may interfere with thyroid hormone synthesis or may exacerbate problems of iodine deficiency. A good example of this is that of perchlorate. This chemical is used as an oxidant in solid rocket propellants, in ordnance, fireworks, airbag deployment systems, and others (322). Because of the environmental stability of perchlorate, it has become a widespread contaminant in drinking and irrigation waters and in food (323), such that perchlorate contamination is nearly ubiquitous in the U.S. population (324). Experimental studies in humans indicate that the serum half-life of perchlorate is about 8 h and that a dose of about 5.2 μg/kg·d is sufficient to begin to reduce iodide uptake into the thyroid gland (325). Thus, it was surprising that Blount et al. (326) found that urinary perchlorate levels were associated with serum TSH in the general population of women (not in men). It is perhaps not surprising that this association was greater in women with urinary iodine below 100 μg/liter and stronger still among these women who smoke (327) because cigarettes contain thiocyanates that also inhibit iodine uptake. Because infants are particularly vulnerable to thyroid hormone insufficiency (328) and because perchlorate levels are particularly high in breast milk (329), it is of concern that perchlorate may be affecting thyroid hormone signaling in early infant development in some proportion of the U.S. population (330). However, several studies have failed to identify such a relationship. For example, Amitai et al. (331) recently reported that newborn T4 levels, taken as part of the newborn screening program, were not different on average in babies born in neighborhoods known to be highly contaminated with perchlorate in drinking water compared with babies born in neighborhoods with lower-level perchlorate contamination. These findings are more consistent with a number of studies employing newborn T4 screening data and location of residence as a proxy measure of perchlorate contamination (for review, see Ref. 322).
There are a number of chemicals that can interfere with iodide uptake by the NIS (332), including chlorate, thiocyanate, and nitrates that are particularly prevalent. It is likely that the effect of one of these chemicals (e.g., perchlorate) on iodide uptake will depend on the presence and concentration of the others and with iodine itself (333).
Iodide, the form of iodine that enters the cell, must be oxidized to a higher oxidation state before it is transferred to the precursor of thyroid hormone, thyroglobulin (334). Of the known biological oxidizing agents, only H2O2 and O2 are capable of oxidizing iodide (335). Organification of iodine is controlled by the enzyme thyroperoxidase (TPO), a heme-containing enzyme. A number of compounds are known to block TPO. A prototypical one is 6-propyl-2-thiouracil (PTU), a methylmercaptoimidazole that has been intensively studied in animals and in humans and is used therapeutically to treat patients with Graves’ disease (336). As a class (the 2-mercapto-4-hydroxy-6-propyl-pyrimidines), PTU is representative of compounds found in the environment that can affect thyroid function. PTU is well known to reduce circulating levels of T4 and T3 and to increase circulating levels of TSH (405) and has been extensively used in mechanistic research focused on identifying the role of thyroid hormone in brain development. The ability of PTU to reduce circulating thyroid hormone levels has been exploited in the treatment of hyperthyroidism in humans, including in pregnant and lactating women (337). PTU is generally believed to produce deleterious effects in animals by causing a dose-dependent reduction in circulating levels of thyroid hormone. This reduction is caused by the ability of PTU to inhibit directly the function of the TPO enzyme (338).
Other TPO inhibitors include the isoflavones, especially those found in soy protein (e.g., genistein, coumesterol; reviewed in Ref. 339). In humans, goiter has been reported in infants fed soy formula (340,341,342). In addition, teenage children diagnosed with autoimmune thyroid disease were found to have twice the rate of occurrence if they had consumed soy formula as infants (343). Boker et al. (344) recently reviewed the dietary sources of a variety of isoflavones, showing that these are common dietary components. These isoflavones are also so-called “phytoestrogens,” which are highly enriched in some commercial preparations.
C. Environmental chemicals impacting thyroid hormone transport, metabolism, and clearance
Once secreted into the blood, thyroid hormones are carried by specific proteins. In humans, about 75% of T4 is bound to T4-binding globulin (TBG), 15–20% is bound to transthyretin (TTR; also called T4 binding prealbumin or TBPA), and the remaining 5–10% is bound to albumin or is free (0.02%) (345,346).
The role of serum binding proteins for thyroid hormone in thyroid homeostasis is not well understood. No single serum T4 binding protein is essential for good health or for the maintenance of a euthyroid state in humans (347). There are a number of clinical situations in which serum binding proteins are elevated or reduced (even completely absent) and the thyroid state is normal. Therefore, despite large increases or decreases in serum total T4 and T3 concentrations in some of these patients, serum free hormone and TSH are normal (348). In contrast, there is evidence that the role of serum binding proteins such as TBG is to allow the equal distribution of hormone delivery to a tissue. Mendel et al. (349) found that 125 I-T4 was evenly distributed in the rodent liver after a single pass through the tissue only if serum binding proteins were present in the perfusate. However, the identity of the serum binding protein (e.g., TTR vs. TBG) did not alter the pattern or intensity of T4 uptake.
There is some evidence that TTR is important in transport of thyroid hormone across the blood-brain barrier. In large part, this concept is derived from the observation that TTR is produced in the choroid plexus (350,351,352). However, this concept is not supported by the observation that mice carrying a targeted deletion of the TTR gene have normal concentrations of T4 in the brain (353,354). A number of chemicals have been shown to displace T4 from TTR in vitro. In fact, some chemicals bind to TTR with higher affinity than does T4 itself (355,356,357); however, the consequences of this binding are not completely clear. One hypothesis is that chemicals can reduce serum total T4 levels by inhibiting T4 binding to TTR (358). Perhaps this displacement may also increase T4 clearance by the liver. However, TTR in the choroid plexus appears to be important for thyroid hormone action in the brain (359), and TTR may mediate transport of environmental chemicals into various compartments such as placenta (360). Thus, chemical binding to the TTR may not only decrease the availability of thyroid hormone to various tissues, it may also selectively target these chemicals for transport and uptake.
Many chemicals are known to decrease the serum half-life of T4 by inducing liver enzymes that glucuronidate T4 (361,362,363). These enzymes uridine diphosphate glucuronyl transferase can be induced by dioxin-like compounds acting on the AhR or through the pregnane X-receptor or constitutive androstane receptor nuclear receptors (364). These chemicals fall into many industrial categories including pesticides of many types (365). The classes of industrial chemicals known to interact with the thyroid system have been reviewed previously and will not be emphasized here (see Refs. 316, 317, 365, and 366).
Once in the serum, thyroid hormones can be taken up into tissues by selective transporters (367) to enter cells. This issue has been particularly investigated because the finding that children with a genetic defect in the MCT8 gene exhibit severe neurological and behavioral disorders (Allan-Herndon-Dudley syndrome; Ref. 368). There are a number of transporters that are likely to be important in the control of thyroid hormone uptake into various tissues and cells. However, little is known—or has been tested—about the ability of specific environmental or industrial chemicals to interfere with T4 or T3 transporter function.
Inside the cell, T4 can be converted to T3 by the type 1 or type 2 deiodinase. These outer-ring deiodinases are essential for thyroid hormone action (369). For example, the type 2 deiodinase knockout mouse exhibits a form of pituitary resistance to thyroid hormone negative feedback in which both serum T4 and TSH are elevated (370), indicating that the conversion of T4 to T3 in pituitary cells is an important step in thyroid hormone action. A number of environmental chemicals affect deiodinase activity including PCBs (360,371,372) and others (317). Environmental chemicals that affect deiodinase activity may have effects that are not entirely consistent with the appearance of “hypothyroidism” and, therefore, may be difficult to recognize in the absence of mechanistic studies.
D. Environmental chemicals impacting the thyroid hormone receptor
1. PCBs
Despite early speculations that environmental chemicals may act as imperfect thyroid hormone analogs (373), few studies had tested this hypothesis until recently. Now, several recent reports show that a broad range of chemicals to which humans are routinely, and inadvertently, exposed can bind to TRs and may produce complex effects on thyroid hormone signaling. Perhaps the best example is that of PCBs—industrial chemicals consisting of paired phenyl rings with various degrees of chlorination (374). Although the production of PCBs was banned in the mid 1970s, these contaminants are routinely detected in the environment (375) and in human tissues (376). PCB body burden is associated with lower full-scale IQ, reduced visual recognition memory, attention deficits, and motor deficits (377,378,379,380,381).
PCBs can reduce circulating levels of T4 in animals (382,383,384), and some authors propose that PCBs exert neurotoxic effects on the developing brain by causing a state of relative hypothyroidism (385,386). In addition, PCB body burden has been found to be associated with thyroid hormone in some, but not all, human studies (366,387). Interestingly, measures of thyroid function at birth are associated with maternal, infant, and delivery factors, and this may explain why some studies fail to identify an association between PCB exposures and measures of thyroid function at birth (388,389).
The concept that PCBs can exert a neurotoxic effect on the developing brain by causing a state of relative hypothyroidism is supported by the observations that the ototoxic effect of PCB exposure in rats can be partially ameliorated by T4 replacement (390), and that the cerebellum, a tissue highly sensitive to thyroid hormone insufficiency (391), is targeted by PCB exposure. PCBs alter motor behavior associated with cerebellar function, as well as cerebellar anatomy (392). Interestingly, PCB exposure is associated with an increase in expression of glial fibrillary acidic protein (392), which is also increased by thyroid hormone insufficiency (393). Finally, in young children, the association between PCB body burden and behavioral measures of response inhibition is stronger in those children that have a smaller corpus callosum (394), an area of the brain affected by thyroid hormone (395). Thus, it is possible that PCBs exert at least some neurotoxic effects on the developing cerebellum by causing a state of relative hypothyroidism.
However, PCB exposure does not produce consistent effects on animals that are indicative of thyroid hormone insufficiency, such as body weight gain during development (382) or the timing of eye opening (390). In addition, despite the reduction in serum T4, PCB exposure increases the expression of several thyroid hormone-responsive genes in the fetal (396,397) and neonatal (382) brain. These observations are consistent with the hypothesis that at least some individual PCB congeners, or their metabolites, can act as TR agonists in vivo. Recently, Kitamura et al. (398) reported that nine separate hydroxylated PCB congeners can bind to the rat TR with an IC50 as low as 5 μ m . In addition, using a human neuroprogenitor cell line, Fritsche et al. (399) found that a specific PCB congener could mimic the ability of T3 in increasing oligodendrocyte differentiation and that this effect was blocked by the selective TR antagonist NH3. Finally, Arulmozhiraja and Morita (400) have identified several PCB congeners that exhibit weak thyroid hormone activity in a yeast two-hybrid assay optimized to identify such activity.
Not all recent reports indicate that PCBs act as agonists on the TR. Kimura-Kuroda et al. (401) have found that two separate hydroxylated PCBs interfere with T3-dependent neurite outgrowth in mouse cerebellar granule cell primary cultures. In addition, Bogazzi et al. (402) found that a commercial mixture of PCBs (Aroclor 1254) exhibited specific binding to the rat TRβ at approximately 10 μ m . This concentration inhibited TR action on the malic enzyme promoter in a chloramphenicol acetyltransferase assay, and this effect required an intact thyroid response element (TRE). However, the PCB mixture did not alter the ability of TR to bind to the malic enzyme TRE in a gel shift assay. In contrast, Iwasaki et al. (403) found that a specific hydroxylated PCB congener inhibits TR-mediated transcriptional activation in a luciferase assay at concentrations as low as 10 −10 m . This effect was observed in several cell lines, but was not observed using a glucocorticoid response element. Miyazaki et al. (404) followed this report by showing that PCBs can dissociate TR:retinoic X receptor (RXR) heterodimers from a TRE.
It is clear that PCBs are neurotoxic in humans and animals and that they can interact directly with the TR. However, the consequences of PCB exposure on TR action appear to be quite complex. This complexity includes acting as an agonist or antagonist and may include TR isoform selectivity inasmuch as most studies have been performed using the TRβ, leaving the TRα relatively unstudied in this context. In addition, considering that there are 209 different chlorine substitution patterns on the biphenyl backbone and that these can be metabolized [hydroxyl and methylsulfonyl metabolites (173,174)], it is possible that different chemical species exert different effects. Finally, PCBs may exert different actions on TRs depending on associated heterodimer partners, promoter structure, or different cofactors. This complexity will be important to pursue because the effect of PCB exposure in humans is far better studied than for structurally related compounds such as PBBs and PBDEs. Thus, mechanistic studies on PCBs can be more easily and effectively coupled to specific human health outcomes.
2. BPA
BPA (4,4′ isopropylidenediphenol) is produced at a rate of more than 800 million kilograms annually in the United States alone (418) and is used primarily in the manufacture of plastics including polycarbonate plastics, epoxy resins that coat food cans, and in dental sealants (406,407). Howe et al. (406) estimated human consumption of BPA from epoxy-lined food cans alone to be about 6.6 μg per person per day. BPA has been reported in concentrations of 1–10 ng/ml in serum of pregnant women, in the amniotic fluid of their fetus, and in cord serum taken at birth (71,408). Moreover, BPA concentrations of up to 100 ng/g were reported in placenta (408). BPA is also halogenated (brominated or chlorinated) to produce flame retardants. Tetrabromobisphenol A (TBBPA) is the most commonly used, with more than 60,000 tons produced annually (409,410). Thomsen et al. (411) recently reported that brominated flame retardants, including TBBPA, have increased in human serum from 1977–1999 with concentrations in adults ranging from 0.4 to 3.3 ng/g serum lipids. However, infants (0–4 yr) exhibited serum concentrations that ranged from 1.6 to 3.5 times higher (411).
Considering this pattern of human exposure, it is potentially important that BPA has been shown to bind to the TR (412). Although best studied for its actions on the nuclear ER (413), binding with a Ki of approximately 10 −5 m (414,415), and more recently for the membrane ER (416), BPA also binds to and antagonizes T3 activation of the TR (412,417) with a Ki of approximately 10 −4 m . As little as 10 −6 m BPA significantly inhibits TR-mediated gene activation (412). Moreover, Moriyama et al. (412) found that BPA reduced T3-mediated gene expression in culture by enhancing the interaction with nuclear receptor corepressors. Interestingly, Zoeller et al. (418) found that developmental exposure to BPA in rats produces an endocrine profile similar to that observed in thyroid resistance syndrome (419). Specifically, T4 levels were elevated during development in the pups of BPA-treated animals, but TSH levels were not different from controls (418). This profile is consistent with BPA inhibition of TRβ-mediated negative feedback. However, the thyroid hormone- response gene RC3 was elevated in the dentate gyrus of these BPA-treated animals (418). Because the TRα isoform is expressed in the dentate gyrus, the authors concluded that BPA could be a selective TRβ antagonist in vivo.
If BPA acts as a TR antagonist in vivo, it is predictable that specific developmental events and behaviors would be affected by developmental exposure to BPA. In this regard, Seiwa et al. (420) have shown that BPA blocks T3-induced oligodendrocyte development from precursor cells. In addition, there may be an association between the thyroid resistance syndrome and attention deficit-hyperactivity disorder in humans (421) and in rats (422); therefore, it is potentially important that BPA-exposed rats exhibit attention deficit-hyperactivity disorder-like symptoms (423).
Despite the antagonistic effects of BPA on the TRβ, halogenated BPAs appear to act as TR agonists (417). Both TBBPA and tetrachlorobisphenol A can bind to the thyroid hormone receptor and induce GH3 cell proliferation and GH production (417). Thus, these compounds may exert agonistic effects on the TR, and this could be important during early brain development. For example, thyroid hormone of maternal origin can regulate gene expression in the fetal brain (424,425,426); one of these genes codes for Hes1 (397). Considering the role of HES proteins in fate specification in the early cortex (427,428), the observation that industrial chemicals can activate the TR and increase HES expression (397) may indicate that these chemicals can exert subtle effects on early differentiative events.
3. PBDEs
PBDEs may also bind to the thyroid hormone receptor (reviewed in Ref. 429). Marsh et al. (430) demonstrated that two hydroxylated PBDEs can bind to both TRα and TRβ, but with a significant preference for TRβ.
IX. Environmental Chemicals, Obesity, and Metabolism
A. Introduction to EDCs and the obesity epidemic
Obesity, defined as body fat greater than 25% in men or greater than 30% in women, is fast becoming a significant human health crisis (431). More than 30% of adults in the United States are defined as clinically obese (431,432), and an analogous rise is observed in pediatric populations, with a tripled increase in the obesity rate from ages 6–19 yr during the last five decades (433). The prevalence of obesity has risen dramatically in wealthy developed countries, and it is also on the rise in poor nations. The WHO has declared excessive weight as one of the top 10 health risks in the world and has estimated that the number of overweight people in the world is now greater than the number of undernourished. The rise in the incidence in obesity matches the rise in the use and distribution of industrial chemicals that may be playing a role in generation of obesity (434), suggesting that EDCs may be linked to this epidemic.
Obesity has deleterious effects on human health by increasing the risk of associated metabolic abnormalities such as insulin resistance, hyperinsulinemia, hypertension, and dislipidemia—all components of the metabolic syndrome—which constitute, in turn, major risk factors for the development of diabetes mellitus type 2 and coronary heart disease. The etiology of the obesity epidemic has been partly attributed to alterations in food intake, with the prevalence of a Westernized-style diet characterized by high caloric uptake as well as a lack of physical activity representative of a sedentary lifestyle. However, the mechanisms still remain unclear, and except for a genetic predisposition and lifestyle modifications, scientific research implies the impact of environmental substances in the generative roots of obesity. Grün and Blumberg (435,436) have coined the terminology “obesogens” in reference to molecules that inappropriately regulate lipid metabolism and adipogenesis to promote obesity.
Obesity also relates to the fetal (developmental) origins of adult disease. Children of women who experienced famine during pregnancy exhibit symptoms of the metabolic syndrome as adults (437). Moreover, it is becoming evident that an important risk factor for development of this metabolic syndrome is low birthweight (438,439). These studies indicate that developmental events occurring in utero and perhaps in the immediate perinatal period can affect metabolic functions that can lead to the metabolic syndrome in adulthood (431).
B. Environmental estrogens and obesity
White adipose tissue metabolism is under the control of the sympathetic nervous system and is modulated by hormones including sex steroids. The impact of environmental estrogens on adipose tissue may be through direct modulation of lipogenesis, lipolysis, and adipogenesis, or indirect by affecting food consumption and leptin secretion targeting the central nervous system or lipid homeostasis in liver (440).
The estrogenic pharmaceutical chemical DES illuminates the relationship between perinatal exposures and latent development of high body weight and obesity. Moreover, there is a complex relationship between the concentration of estrogen to which pregnant animals are exposed and the weight of the offspring in adulthood (432). Specifically, according to a recent experiment by Newbold et al. (432), mice neonatally exposed to DES experience increased body weight in adulthood associated with excess abdominal body fat. Interestingly, the dose of DES determines the chronic manifestation of the observed alterations, with high doses leading to initially decreased body weight and a peripubertal “catch-up” and low doses causing an increase in weight detectable only in adulthood. Moreover, the timing is important because gestational administration in rodents results in the offspring’s low birth weight, an unchanged metabolic characteristic throughout life (432). Along with an increase in body fat stores, the adipokines leptin and adiponectin, IL-6 (an inflammatory marker), and triglycerides were all elevated in DES-exposed mice (432).
An in vitro study using a culture system of 3T3-L1 preadipocytes showed that 4-nonylphenol and BPA stimulated lipid accumulation, accelerating their differentiation to mature adipocytes in a time- and concentration-dependent way (441). The underlying mechanism appeared to involve up-regulation of gene expression involved in lipid metabolism and adipocyte differentiation. In the second part of the experiment, fat accumulation was observed in human hepatocellular carcinoma cell lines exposed to those endocrine disruptors (441). These findings are consistent with previous in vitro studies using mouse fibroblast cell lines in which a link between environmental chemicals including nonylphenol, BPA, and genistein in the development of body weight imbalance was suggested (431,432).
C. Peroxisome proliferator-activated receptor (PPAR) γ and organotins
PPARγ is a member of the nuclear receptor superfamily and constitutes a major regulator of adipogenesis. It is primarily expressed in adipose tissue, and its activation promotes adipocyte differentiation as well as the induction of lipogenic enzymes. Additionally, it contributes to maintenance of metabolic homeostasis through transcriptional activation of genes implicated in energy balance (442). During its activation, PPARγ forms a heterodimer with RXR-α, and the complex binds to PPAR response elements in the regulatory regions (promoters) of target genes ultimately involved in the regulation of fatty acid storage and the repression of lipolysis.
Experimental evidence highlights that nuclear receptor superfamily and specifically PPARγ are molecular targets for endocrine disruptors, in particular organotin compounds such as tributyltin (TBT) and triphenyltin, which have been widely used in agriculture and industry. Kanayama et al. (443) showed that TBT and triphenyltin functioned as agonists of PPARγ and RXR, acting as high-affinity ligands at levels comparable to known endogenous ligands. Moreover, administration of those xenobiotics in preadipocyte cell lines resulted in adipocyte differentiation through PPARγ (443). In mice, TBT induced the differentiation of adipocytes in vitro and increased adipose mass in vivo by RXR and PPARγ activation (444).
It is possible that PPARγ signaling can interact with that of estrogen to influence adipogenesis. These findings have been reviewed recently (435,436,444) and represent an important example of the mechanism by which environmental chemicals can interfere with body weight regulation. In addition, at high doses, TBT can inhibit aromatase enzyme activity in adipose tissue directly, leading to decreased estradiol levels and down-regulation of ER target genes. TBT at moderate to high doses inhibits the activity of 11β-hydroxysteroid dehydrogenase 2, resulting in decreased inactivation of cortisol. It has been hypothesized that the increased local glucocorticoid levels could influence late stages in adipocyte differentiation and thus, metabolic regulation (435,436).
D. Phytoestrogens
In recent years, efforts to implement healthier eating habits have resulted in an increased consumption of soy products and supplements and hence, increased exposure to phytoestrogens. Genistein is the principal phytoestrogen in soy and has a wide range of biological actions. It binds to ERα and ERβ but also displays antiestrogenic action (445). At low concentrations, genistein was found to act as estrogen and exert an inhibitory effect on lipogenesis. There are also sex differences in the effect of genistein on adipose deposition and insulin resistance, an effect that involves the ERβ (446). At higher concentrations, genistein promotes lipogenesis through the molecular pathway of PPARγ, an ΕR-independent pathway (445).
E. Endocrine disruptors, diabetes, and glucose homeostasis
The incidence of diabetes mellitus has tripled over recent decades, with an estimated 177 million people affected worldwide (447). It is speculated that by the year 2030 the prevalence of diabetes will increase to 4.4% worldwide (from 2.8% in 2000) with more than 300 million diabetic adults (448). Regarding the young population, epidemiological studies show an alarming increase in the incidence of diabetes mellitus type 2 (449).
Based on the links between endocrine disruptors and disturbances of reproduction, metabolism, and links to adult dysfunctions and cancer, it is reasonable to propose a connection between EDCs and diabetes as well as prediabetic disturbances. Indeed, epidemiological studies have linked high dioxin levels with increased risk for diabetes or altered glucose metabolism (450). Animal models also support this hypothesis. Alonso-Magdalena et al. (447) undertook an in vivo experiment to evaluate the impact of BPA on pancreatic β-cell function. Its biological action was compared with 17β-estradiol. The results showed that acute treatments with either estradiol or BPA caused a temporary hyperinsulinemia, whereas longer-term exposure provoked insulin resistance with chronic increased insulin levels, an aggravating factor for the development of diabetes mellitus (447). Recently, in conditioned media from human breast, sc and visceral adipose explants, it was demonstrated that BPA at environmentally relevant doses (0.1 and 1 n m ) inhibits the release of adiponectin, an adipocyte-specific hormone that increases insulin sensitivity. Therefore, factors that suppress adiponectin release could aggravate insulin resistance and susceptibility to obesity-related syndromes like metabolic syndrome and type 2 diabetes mellitus. However, the mechanisms by which BPA suppresses adiponectin and the receptors involved remain to be determined (451).
Pancreatic α-cells have also been suggested as potential targets for endocrine disruption. Low doses of BPA and DES were shown to impair the molecular signaling that leads to secretion of glucagon by suppressing intracellular calcium ion oscillations in α-cells in response to low blood glucose levels through a nongenomic mechanism (452).
The above experiments suggest that low doses of endocrine disruptors can disrupt pancreatic physiology, affecting both insulin- and glucagon-secretory cells, leading to changes in the regulation of glucose and lipid metabolism. The underlying mechanisms involve at the very least classical ER-mediated but also nongenomic actions. Further investigations are required to elucidate the potential associations with human health. Importantly, whereas current evidence represents experimental data from laboratory animals or in vitro studies, no direct association with humans has yet been established, with the exception of the epidemiological studies discussed above.
F. Endocrine disruptors and cardiovascular systems
The obesity phenotype may lead to a dysmetabolic state with atherogenic, inflammatory, prothrombotic abnormalities that not only accelerate the progression of cardiovascular disease but also create favorable “subsoil” for an acute myocardial infarct (453). Therefore, the cardiovascular system is also a target of environmental chemicals that interfere with intracellular signaling of hormonal and inflammatory pathways.
G. Estrogenic EDCs and cardioprotection
Phytoestrogens have been shown to exert cardioprotective effects. Female rats fed a high phytoestrogen diet exhibited cardioprotection against adverse left ventricular remodeling (454) and reduction of myocardial necrosis, increased myocardial contractility, and decreased occurrence of ventricular arrhythmias. Genistein was also associated with reduced levels of TNF-α and blunted myocardial intercellular adhesion molecule-1 expression (455). Moreover, it was shown that in people at high risk of cardiovascular events, a greater isoflavone intake is associated with better vascular endothelial function and lower carotid atherosclerotic burden (456).
Regarding human populations, there are some epidemiological studies that suggest that high phytoestrogen intake is inversely associated with cardiovascular risk factors and development of cardiovascular disease (457). Moreover, it was shown that in people at high risk of cardiovascular events, a greater isoflavone intake is associated with better vascular endothelial function and lower carotid atherosclerotic burden (456). However, these epidemiological observations need clinical confirmation.
H. Advanced glycation end-products (AGEs)
Recent data clearly suggest that a heterogeneous group of exogenous advanced glycation end-products (AGEs) have a negative impact on cardiometabolic tissues. Tobacco use (458) and food cooked at high temperatures, precooked meals, and some beverages contain large amounts of AGEs that are absorbed from the human gastrointestinal tract (459). AGEs cause tissue injury through intracellular generation of free radicals and triggering oxidative stress, through the interaction of AGEs with a multiligand, cell surface receptor called RAGE, and endocrine signaling pathway. There is evidence in experimental animals and humans for a link between exogenous AGEs and an increase in cardiometabolic risk markers. It is notable that mice chronically fed a high-AGE diet, compared with those fed a low-AGE, high-fat diet exhibited relative insulin resistance accompanied by modifications in pancreatic cellular architecture compatible with hyperplasia and hypertrophy and loss of islet of Langerhans structure (460). Another in vivo chronic experiment involved feeding intact female rats a high-AGE diet for 6 months. This resulted in increased fasting glucose and insulin levels independent of the degree of obesity as well as hormonal alterations (461). Uribarri et al. (462) showed that a single oral administration of an AGE-rich beverage acutely (within 90 min) resulted in temporarily impaired endothelial function assessed by flow-mediated arterial vasodilation, increased serum C-reactive protein, and plasminogen activator inhibitor-1 levels in both diabetic and healthy subjects.
I. Conclusions
The literature demonstrates a role of EDCs in the etiology of complex diseases such as obesity, diabetes mellitus, and cardiovascular disease, yet these processes are still poorly understood. Although the evidence is limited, accumulating data are pointing to the potential role of endocrine disruptors either directly or indirectly in the pathogenesis of adipogenesis and diabetes, the major epidemics of the modern world. Taking into consideration the wide spectrum of industrial chemicals to which an average consumer might be exposed, a rational hypothesis is that the scientific community may inadvertently ignore the effect of several other compounds that might in turn constitute potential “obesogens” or promoters of glycemic disturbances. Further research is required to elucidate all potential interactions between environmental substances and metabolic dysregulation.
X. Recommendations for the Future
A. Linking basic research to clinical practice
It should be clear from this Scientific Statement that there is considerable work to be done. A reconciliation of the basic experimental data with observations in humans needs to be achieved through translation in both directions, from bench to bedside and from bedside (and populations) to bench. An example of how human observation and basic research have successfully converged was provided by DES exposure in humans, which revealed that the human syndrome is faithfully replicated in rodent models. Furthermore, we now know that DES exposure in key developmental life stages can have a spectrum of effects spanning female reproduction, male reproduction, obesity, and breast cancer. It is interesting that in the case of breast cancer, an increased incidence is being reported now that the DES human cohort is reaching the age of breast cancer prevalence. The mouse model predicted this outcome 25 yr before the human data became available. In the case of reproductive cancers, the human and mouse data have since been confirmed in rats, hamsters, and monkeys (463). This is a compelling story from the perspective of both animal models and human exposures on the developmental basis of adult endocrine disease.
Another estrogenic compound, BPA, is also linked to a wide variety of endocrine dysfunction. BPA exposure, particularly in development, increases the risk of mammary cancer, obesity, diabetes, and reproductive and neuroendocrine disorders. The human evidence for BPA is mounting; recently, Lang et al. (464) published a cross-sectional analysis on the relationship between concentrations of urinary BPA and chronic disease states in over 1400 adults in the United States. They found a significant correlation between BPA concentrations in urine with cardiovascular disease and abnormal concentrations of liver enzymes. It would be really interesting to be able to relate the relationship of these outcomes with developmental/fetal exposure to BPA and other xenobiotics. However, epidemiological research on fetal exposure would be logistically difficult and costly because exposures must be measured at several different time points, including gestation, whereas the outcome may not be manifest in some cases until 50 or more years after the initial fetal exposure. Given the reproducibility of the human DES syndrome in rodents and recent evidence for commonalities in a relationship between BPA and cardiovascular endocrine disease, it is obvious that more research in animal models is necessary to enrich our knowledge of the mechanisms by which endocrine disruptors increase the risk of disease.
A challenge to understanding the relationship between EDCs and health abnormalities is that EDCs are a “moving target.” Individuals and populations are exposed to ever-changing patterns of production and use of these compounds. They also tend to be released into the environment as mixtures, rather than individual chemicals. Therefore, it is important to understand the effects of simultaneous coexposures to these chemicals, which may interact additively, multiplicatively (synergistically), or antagonistically (48). There are limited data on the interactions between chemicals within a class or across classes of chemicals. Presently, there are good analytical methods for measuring exposures to a variety of endocrine disruptors in humans. An increased understanding of the potential human health risks of exposure to mixtures of EDC is important but remains very understudied. Hence, measurement of body burden of the most prevalent xenobiotics would probably be the best strategy for finding a link between exposure and effect. Once known, this could be related to mechanistic studies in laboratory models, and future experiments could be designed to evaluate the effects of combinations of common EDCs in the laboratory, with the obvious caveat that it will not be possible to mimic every possible combination and dose. Despite these challenges, evolving and innovative technologies designed to improve the assessment of human exposure and reproductive and endocrine health endpoints should provide enhanced opportunities for improving our understanding of these relationships.
B. Endocrine disruption and the public
At the recent Summit on Environmental Challenges to Reproductive Health and Fertility at the University of California, San Francisco, recommendations were made regarding future research, health care, policy, community action, and occupational health (49). Included in these recommendations were enhancing collaborations among and between researchers and granting agencies and promoting critical research directions, including prenatal exposures in the National Children’s Health Study, leveraging specific laboratory data into the National Health and Nutrition Examination Survey study, developing biomarkers of exposure and disease, and increasing the funding for effects of chemicals on the epigenome, developmental programming, transgenerational effects, and cross-talk among endocrine systems and metabolic and immune systems (49). In addition, for health care professionals, being educated in sources and effects of environmental contaminant exposures in utero and across the life span, as well as having straightforward health information tools to share this information with patients and for public education in general are recommended.
C. Prevention and the “precautionary principle”
Although more experiments are being performed to find the hows and whys, what should be done to protect humans? The key to minimizing morbidity is preventing the disorders in the first place. However, recommendations for prevention are difficult to make because exposure to one chemical at a given time rarely reflects the current exposure history or ongoing risks of humans during development or at other life stages, and we usually do not know what exposures an individual has had in utero or in other life stages.
In the absence of direct information regarding cause and effect, the precautionary principle is critical to enhancing reproductive and endocrine health (49). As endocrinologists, we suggest that The Endocrine Society actively engages in lobbying for regulation seeking to decrease human exposure to the many endocrine-disrupting agents. Scientific societies should also partner to pool their intellectual resources and to increase the ranks of experts with knowledge about EDCs who can communicate to other researchers, clinicians, community advocates, and politicians.
D. Specific recommendations for future research
Although direct causal links between exposures to EDCs and disease states in humans are difficult to draw, results from basic research and epidemiological studies make it clear that more screening for exposures and targeting at-risk groups is a high priority. In addition to enhancing research in these areas, an important and effective approach is prevention of disease. Our chemical policies at local, state, and national levels, as well as globally, need to be formulated, financed, and implemented to ensure the best public health. Additional specific recommendations of this group are shown in Box 2. By communicating these priorities to basic and clinical researchers, physicians, community advocates, and the public at large, we are hopeful that early identification and intervention will be facilitated.
Acknowledgments
We thank Dr. Loretta Doan for the critical role that she played in facilitating all communications of this taskforce; Dr. Alan Schneyer for support and guidance; Ms. Maggie Haworth for expert assistance with publication-related activities; the Research Affairs Core Committee of The Endocrine Society; and The Scientific Statements Taskforce of The Endocrine Society.
Footnotes
This work was supported by Grants NIH ES07784 (to A.C.G.), NIH ES015584 (to G.S.P.), NIH ES09718 (to R.H.), NIH ES01230, ES015182, ES08314 (to A.M.S.), European Commission (EDEN project, QLRT-2001-00269), Belgian Study Group for Pediatric Endocrinology, Belgian Fonds de la Recherche Scientific Medicale, grants 3.4567.09 and 3.4573.05 (to J.P.B.).
Disclosure Summary: E.D.-K., J.-P.B., L.C.G., R.H., G.S.P., and A.C.G. have nothing to declare. A.M.S. has served as a consultant to Emerson Poynter LLP. R.T.Z. has served on the EPA Scientific Advisory Board, has received honoraria for university lectures, and has served as an expert witness in federal court.
Abbreviations: AGE, Advanced glycation end-product; AhR, aryl hydrocarbon receptor; AR, androgen receptor; BPA, bisphenol A; DDE, dichlorodiphenyldichloroethylene; DDT, dichlorodiphenyltrichloroethane; DES, diethylstilbestrol; DMBA, dimethylbenzanthracene; EDC, endocrine-disrupting compound; ER, estrogen receptor; HPA, hypothalamic-pituitary-adrenal axis; HPG, hypothalamic-pituitary-gonadal axis; HPT, hypothalamic-pituitary-thyroid axis; IUGR, intrauterine growth retardation; IVF, in vitro fertilization; MBP, monobutyl phthalate; MBzP, monobenzyl phthalate; NIS, sodium/iodide symporter; PBB, polybrominated biphenyl; PBDE, polybrominated diphenyl ether; PCB, polychlorinated biphenyl; PCOS, polycystic ovarian syndrome; POF, premature ovarian failure; PPARγ, peroxisome proliferator-activated receptor γ; PTU, 6-propyl-2-thiouracil; RXR, retinoic X receptor; TBBPA, tetrabromobisphenol-A; TBG, T4-binding globulin; TBT, tributyltin; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; TDS, testicular dysgenesis syndrome; TGCC, testicular germ cell cancer; TPO, thyroperoxidase; TR, thyroid receptor; TRE, thyroid response element; TTR, transthyretin.
References
- Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA 1998 Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology 139:4252–4263 [PubMed] [Google Scholar]
- Dickerson SM, Gore AC 2007 Estrogenic environmental endocrine-disrupting chemical effects on reproductive neuroendocrine function and dysfunction across the life cycle. Rev Endocr Metab Disord 8:143–159 [PubMed] [Google Scholar]
- Cao Y, Calafat AM, Doerge DR, Umbach DM, Bernbaum JC, Twaddle NC, Ye X, Rogan WJ 2009 Isoflavones in urine, saliva and blood of infants—data from a pilot study on the estrogenic activity of soy formula. J Expo Sci Environ Epidemiol 19:223–234 [PMC free article] [PubMed] [Google Scholar]
- Calafat AM, Needham LL 2007 Human exposures and body burdens of endocrine-disrupting chemicals. In: Gore AC, ed. Endocrine-disrupting chemicals: from basic research to clinical practice. Totowa, NJ: Humana Press; 253–268 [Google Scholar]
- Porte C, Janer G, Lorusso LC, Ortiz-Zarragoitia M, Cajaraville MP, Fossi MC, Canesi L 2006 Endocrine disruptors in marine organisms: approaches and perspectives. Comp Biochem Physiol C Toxicol Pharmacol 143:303–315 [PubMed] [Google Scholar]
- Stahlhut RW, Welshons WV, Swan SH 2009 Bisphenol A data in NHANES suggest longer than expected half-life, substantial non-food exposure, or both. Environ Health Perspect 117:784–789 [PMC free article] [PubMed] [Google Scholar]
- Gore AC, Crews D 2009 Environmental endocrine disruption of brain and behavior. In: Pfaff DW, Arnold AP, Etgen A, Fahrbach S, Rubin R, eds. Hormones, Brain and Behavior. San Diego, Academic Press, pp. 1789–1816 [Google Scholar]
- Barker DJP 2003 The developmental origins of adult disease. Eur J Epidemiol 18:733–736 [PubMed] [Google Scholar]
- Crews D, Putz O, Thomas P, Hayes T, Howdeshell K 2003 Animal models for the study of the effects of mixtures, low doses, and the embryonic environment on the action of endocrine disrupting chemicals. Pure and Applied Chemistry, SCOPE/IUPAC Project Implications of Endocrine Active Substances for Humans and Wildlife 75:2305–2320 [Google Scholar]
- Sheehan DM, Willingham EJ, Bergeron JM, Osborn CT, Crews D 1999 No threshold dose for estradiol-induced sex reversal of turtle embryos: how little is too much? Environ Health Perspect 107:155–159 [PMC free article] [PubMed] [Google Scholar]
- vom Saal FS, Akingbemi BT, Belcher SM, Birnbaum LS, Crain DA, Eriksen M, Farabollini F, Guillette Jr LJ, Hauser R, Heindel JJ, Ho SM, Hunt PA, Iguchi T, Jobling S, Kanno J, Keri RA, Knudsen KE, Laufer H, LeBlanc GA, Marcus M, McLachlan JA, Myers JP, Nadal A, Newbold RR, Olea N, Prins GS, Richter CA, Rubin BS, Sonnenschein C, Soto AM, Talsness CE, Vandenbergh JG, Vandenberg LN, Walser-Kuntz DR, Watson CS, Welshons WV, Wetherill Y, Zoeller RT 2007 Chapel Hill Bisphenol A Expert Panel Consensus Statement: integration of mechanisms, effects in animals and potential to impact human health at current levels of exposure. Reprod Toxicol 24:131–138 [PMC free article] [PubMed] [Google Scholar]
- Anway MD, Skinner MK 2006 Epigenetic transgenerational actions of endocrine disruptors. Endocrinology 147:S43–S49 [PubMed] [Google Scholar]
- Thornton JW 2001 Evolution of vertebrate steroid receptors from an ancestral estrogen receptor by ligand exploitation and serial genome expansions. Proc Natl Acad Sci USA 98:5671–5676 [PMC free article] [PubMed] [Google Scholar]
- Bay K, Asklund C, Skakkebaek NE, Andersson AM 2006 Testicular dysgenesis syndrome: possible role of endocrine disrupters. Best Pract Res Clin Endocrinol Metab 20:77–90 [PubMed] [Google Scholar]
- Toppari J, Kaleva M, Virtanen HE 2001 Trends in the incidence of cryptorchidism and hypospadias, and methodological limitations of registry-based data. Hum Reprod Update 7:282–286 [PubMed] [Google Scholar]
- Hemminki K, Li X 2002 Cancer risks in second-generation immigrants to Sweden. Int J Cancer 99:229–237 [PubMed] [Google Scholar]
- Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP 2003 The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends and changes after migration. Endocr Rev 24:668–693 [PubMed] [Google Scholar]
- Ibáñez L, de Zegher F 2006 Puberty and prenatal growth. Mol Cell Endocrinol 254–255:22–25 [PubMed] [Google Scholar]
- Herbst AL, Ulfelder H, Poskanzer DC 1971 Adenocarcinoma of vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med 284:878–881 [PubMed] [Google Scholar]
- Sharpe RM 2006 Pathways of endocrine disruption during male sexual differentiation and masculinisation. Best Pract Res Clin Endocrinol Metab 20:91–110 [PubMed] [Google Scholar]
- Skakkebaek NE, Rajpert-De Meyts E, Main KM 2001 Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects. Hum Reprod 16:972–978 [PubMed] [Google Scholar]
- Gray Jr LE, Ostby J, Furr J, Price M, Veeramachaneni DN, Parks L 2000 Perinatal exposure to the phthalates DEHP, BBP, and DINP, but not DEP, DMP, or DOTP, alters sexual differentiation of the male rat. Toxicol Sci 58:350–365 [PubMed] [Google Scholar]
- Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM, Mao CS, Redmon JB, Ternand CL, Sullivan S, Teague JL 2005 Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect 113:1056–1061 [PMC free article] [PubMed] [Google Scholar]
- Maffini M, Rubin B, Sonnenschein C, Soto A 2006 Endocrine disruptors and reproductive health: the case of bisphenol-A. Mol Cell Endocrinol 254–255:179–186 [PubMed] [Google Scholar]
- Buck Louis GM, Gray Jr LE, Marcus M, Ojeda SR, Pescovitz OH, Witchel SF, Sippell W, Abbott DH, Soto A, Tyl RW, Bourguignon JP, Skakkebaek NE, Swan SH, Golub MS, Wabitsch M, Toppari J, Euling SY 2008 Environmental factors and puberty timing: expert panel research needs. Pediatrics 121:S192–S207 [PubMed] [Google Scholar]
- Colón I, Caro D, Bourdony CJ, Rosario O 2000 Identification of phthalate esters in the serum of young Puerto Rican girls with premature breast development. Environ Health Perspect 108:895–900 [PMC free article] [PubMed] [Google Scholar]
- Rasier G, Parent AS, Gérard A, Lebrethon MC, Bourguignon JP 2007 Early maturation of gonadotropin-releasing hormone secretion and sexual precocity after exposure of infantile female rats to estradiol or dichlorodiphenyltrichloroethane. Biol Reprod 77:734–742 [PubMed] [Google Scholar]
- Li S, Hursting SD, Davis BJ, McLachlan JA, Barrett JC 2003 Environmental exposure, DNA methylation and gene regulation. Lessons from diethylstilbestrol-induced cancers. Ann NY Acad Sci 983:161–169 [PubMed] [Google Scholar]
- McLachlan JA, Simpson E, Martin M 2006 Endocrine disrupters and female reproductive health. Best Pract Res Clin Endocrinol Metab 20:63–75 [PubMed] [Google Scholar]
- Darbre PD 2006 Environmental oestrogens, cosmetics and breast cancer. Best Pract Res Clin Endocrinol Metab 20:121–143 [PubMed] [Google Scholar]
- Fenton SE 2006 Endocrine-disrupting compounds and mammary gland development: Early exposure and later life consequences. Endocrinology 147:S18–S24 [PubMed] [Google Scholar]
- Newbold RR, Jefferson WN, Padilla-Banks E 2007 Long-term adverse effects of neonatal exposure to bisphenol A on the murine female reproductive tract. Reprod Toxicol 24:253–258 [PMC free article] [PubMed] [Google Scholar]
- Crews D, McLachlan JA 2006 Epigenetics, evolution, endocrine disruption, health, and disease. Endocrinology 147:S4–S10 [PubMed] [Google Scholar]
- Anway MD, Skinner MK 2008 Transgenerational effects of the endocrine disruptor vinclozolin on the prostate transcriptome and adult onset disease. Prostate 68:517–529 [PMC free article] [PubMed] [Google Scholar]
- Anway MD, Cupp AS, Uzumcu M, Skinner MK 2005 Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308:1466–1469 [PubMed] [Google Scholar]
- Christiansen S, Scholze M, Axelstad M, Boberg J, Kortenkamp A, Hass U 2008 Combined exposure to anti-androgens causes markedly increased frequencies of hypospadias in the rat. Int J Androl 31:241–248 [PubMed] [Google Scholar]
- Shono T, Suita S, Kai H, Yamaguchi Y 2004 Short-time exposure to vinclozolin in utero induces testicular maldescent associated with a spinal nucleus alteration of the genitofemoral nerve in rat. J Pediatr Surg 39:217–219 [PubMed] [Google Scholar]
- Monosson E, Kelce WR, Lambright C, Ostby J, Gray Jr LE 1999 Peripubertal exposure to the antiandrogenic fungicide, vinclozolin, delays puberty, inhibits the development of androgen-dependent tissues, and alters androgen receptor function in the male rat. Toxicol Ind Health 15:65–79 [PubMed] [Google Scholar]
- Newbold RR, Hanson RB, Jefferson WN, Bullock BC, Haseman J, McLachlan JA 1998 Increased tumors but uncompromised fertility in the female descendants of mice exposed developmentally to diethylstilbestrol. Carcinogenesis 19:1655–1663 [PubMed] [Google Scholar]
- Kadlubar FF, Berkowitz GS, Delongchamp RR, Wang C, Green BL, Tang G, Lamba J, Schuetz E, Wolff MS 2003 The CYP3A4*1B variant is related to the onset of puberty, a known risk factor for the development of breast cancer. Cancer Epidemiol Biomarkers Prev 12:327–331 [PubMed] [Google Scholar]
- Rivas A, Fisher JS, McKinnell C, Atanassova N, Sharpe RM 2002 Induction of reproductive tract developmental abnormalities in the male rat by lowering androgen production or action in combination with a low dose of diethylstilbestrol: evidence for importance of the androgen-estrogen balance. Endocrinology 143:4797–4808 [PubMed] [Google Scholar]
- Ceccarelli I, Della Seta D, Fiorenzani P, Farabollini F, Aloisi AM 2007 Estrogenic chemicals at puberty change ERα in the hypothalamus of male and female rats. Neurotoxicol Teratol 29:108–115 [PubMed] [Google Scholar]
- Atanassova N, McKinnell C, Williams K, Turner KJ, Fisher JS, Saunders PT, Millar MR, Sharpe RM 2001 Age-, cell- and region- specific immunoexpression of estrogen receptor α (but not estrogen receptor β) during postnatal development of the epididymis and vas deferens of the rat and disruption of this pattern by neonatal treatment with diethylstilbestrol. Endocrinology 142:874–886 [PubMed] [Google Scholar]
- Khurana S, Ranmal S, Ben-Jonathan N 2000 Exposure of newborn male and female rats to environmental estrogens: delayed and sustained hyperprolactinemia and alterations in estrogen receptor expression. Endocrinology 141:4512–4517 [PubMed] [Google Scholar]
- Kester MH, Bulduk S, van Toor H, Tibboel D, Meinl W, Glatt H, Falany CN, Coughtrie MW, Schuur AG, Brouwer A, Visser TJ 2002 Potent inhibition of estrogen sulfotransferase by hydroxylated metabolites of polyhalogenated aromatic hydrocarbons reveals alternative mechanism for estrogenic activity of endocrine disrupters. J Clin Endocrinol Metab 87:1142–1150 [PubMed] [Google Scholar]
- Waring RH, Harris RM 2005 Endocrine disrupters: a human risk? Mol Cell Endocrinol 244:2–9 [PubMed] [Google Scholar]
- Odum J, Tinwell H, Tobin G, Ashby J 2004 Cumulative dietary energy intake determines the onset of puberty in female rats. Environ Health Perspect 112:1472–1480 [PMC free article] [PubMed] [Google Scholar]
- Kortenkamp A 2008 Low dose mixture effects of endocrine disrupters: implications for risk assessment and epidemiology. Int J Androl 31:233–240 [PubMed] [Google Scholar]
- Woodruff TJ, Carlson A, Schwartz JM, Giudice LC 2008 Proceedings of the Summit on Environmental Challenges to Reproductive Health and Fertility: Executive Summary. Fertil Steril 89:281–300 [PMC free article] [PubMed] [Google Scholar]
- Woodruff TK, Walker CL 2008 Fetal and early postnatal environmental exposures and reproductive health effects in the female. Fertil Steril 89:e47–e51 [PMC free article] [PubMed] [Google Scholar]
- Caserta D, Maranghi L, Mantovani A, Marci R, Maranghi F, Moscarini M 2008 Impact of endocrine disruptor chemicals in gynaecology. Hum Reprod Update 14:59–72 [PubMed] [Google Scholar]
- Crain DA, Janssen SJ, Edwards TM, Heindel J, Ho SM, Hunt P, Iguchi T, Juul A, McLachlan JA, Schwartz J, Skakkebaek N, Soto AM, Swan S, Walker C, Woodruff TK, Woodruff TJ, Giudice LC, Guillette Jr LJ 2008 Female reproductive disorders: the roles of endocrine-disrupting compounds and developmental timing. Fertil Steril 90:911–940 [PMC free article] [PubMed] [Google Scholar]
- Foster WG, Neal MS, Han MS, Dominguez MM 2008 Environmental contaminants and human infertility: hypothesis or cause for concern. J Toxicol Environ Health B Crit Rev 11:162–176 [PubMed] [Google Scholar]
- Mendola P, Messer LC, Rappazzo K 2008 Science linking environmental contaminant exposures with fertility and reproductive health impacts in the adult female. Fertil Steril 89:e81–e94 [PubMed] [Google Scholar]
- The Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group 2004 Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 19:41–47 [PubMed] [Google Scholar]
- Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, Janssen OE, Legro RS, Norman RJ, Taylor AE, Witchel SF 2006 Position statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome. An Androgen Excess Society guideline. J Clin Endocrinol Metab 91:4237–4245 [PubMed] [Google Scholar]
- Legro RS, Azziz R, Giudice L 2006 A twenty-first century research agenda for polycystic ovary syndrome. Best Pract Res Clin Endocrinol Metab 20:331–336 [PubMed] [Google Scholar]
- Franks S, Mason H, Willis D 2000 Follicular dynamics in the polycystic ovary syndrome. Mol Cell Endocrinol 163:49–52 [PubMed] [Google Scholar]
- Yildiz BO, Knochenhauer ES, Azziz R 2008 Impact of obesity on the risk for polycystic ovary syndrome. J Clin Endocrinol Metab 93:162–168 [PMC free article] [PubMed] [Google Scholar]
- Dumesic DA, Abbott DH, Padmanabhan V 2007 Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord 8:127–141 [PMC free article] [PubMed] [Google Scholar]
- Abbott DH, Barnett DK, Bruns CM, Dumesic DA 2005 Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update 11:357–374 [PubMed] [Google Scholar]
- West C, Foster DL, Evans NP, Robinson J, Padmanabhan V 2001 Intra-follicular activin availability is altered in prenatally-androgenized lambs. Mol Cell Endocrinol 185: 51–59 [PubMed] [Google Scholar]
- Chen J, Ahn KC, Gee NA, Ahmed MI, Duleba AJ, Zhao L, Gee SJ, Hammock BD, Lasley BL 2008 Triclocarban enhances testosterone action: a new type of endocrine disruptor? Endocrinology 149:1173–1179 [PMC free article] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Piperi C 2005 Genetics of polycystic ovary syndrome: searching for the way out of the labyrinth. Hum Reprod Update 11:631–643 [PubMed] [Google Scholar]
- Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO 2004 The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab 89:2745–2749 [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Kouli CR, Bergiele AT, Filandra FA, Tsianateli TC, Spina GG, Zapanti ED, Bartzis MI 1999 A survey of the polycystic ovary syndrome in the Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab 84:4006–4011 [PubMed] [Google Scholar]
- Asunción M, Calvo RM, San Millán JL, Sancho J, Avila S, Escobar-Morreale HF 2000 A prospective study of the prevalence of the polycystic ovary syndrome in unselected Caucasian women from Spain. J Clin Endocrinol Metab 85:2434–2438 [PubMed] [Google Scholar]
- Azziz R, Marin C, Hoq L, Badamgarav E, Song P 2005 Health care-related economic burden of the polycystic ovary syndrome during the reproductive life span. J Clin Endocrinol Metab 90:4650–4658 [PubMed] [Google Scholar]
- Takeuchi T, Tsutsumi O, Ikezuki Y, Takai Y, Taketani Y 2004 Positive relationship between androgen and the endocrine disruptor, bisphenol A, in normal women and women with ovarian dysfunction. Endocr J 51:165–169 [PubMed] [Google Scholar]
- Takeuchi T, Tsutsumi O, Ikezuki Y, Kamei Y, Osuga Y, Fujiwara T, Takai Y, Momoeda M, Yano T, Taketani Y 2006 Elevated serum bisphenol A levels under hyperandrogenic conditions may be caused by decreased UDP-glucuronsyltransferase activity. Endocr J 53:485–491 [PubMed] [Google Scholar]
- Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y 2002 Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod 17:2839–2841 [PubMed] [Google Scholar]
- Sinha P, Kuruba N 2007 Premature ovarian failure. J Obstet Gynaecol 27:16–19 [PubMed] [Google Scholar]
- McNatty KP, Reader K, Smith P, Heath DA, Juengel JL 2007 Control of ovarian follicular development to the gonadotrophin-dependent phase: a 2006 perspective. Soc Reprod Fertil Suppl 64:55–68 [PubMed] [Google Scholar]
- Kaipia A, Hsueh AJ 1997 Regulation of ovarian follicle atresia. Annu Rev Physiol 59:349–363 [PubMed] [Google Scholar]
- Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, Du C, Tang W, HämäläinenT, Peng SL, Lan ZJ, Cooney AJ, Huhtaniemi I, Liu K 2008 Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science 319:611–613 [PubMed] [Google Scholar]
- Hunt PA, Koehler KE, Susiarjo M, Hodges CA, Ilagan A, Voigt RC, Thomas S, Thomas BF, Hassold TJ 2003 Bisphenol A exposure causes meiotic aneuploidy in the female mouse. Curr Biol 13:546–553 [PubMed] [Google Scholar]
- Susiarjo M, Hassold TJ, Freeman E, Hunt PA 2007 Bisphenol A exposure in utero disrupts early oogenesis in the mouse. PLoS Genet 3:e5 [PMC free article] [PubMed] [Google Scholar]
- McLachlan JA, Newbold RR, Shah HC, Hogan MD, Dixon RL 1982 Reduced fertility in female mice exposed transplacentally to diethylstilbestrol (DES). Fertil Steril 38:364–371 [PubMed] [Google Scholar]
- Hatch EE, Troisi R, Wise LA, Hyer M, Palmer JR, Titus-Ernstoff L, Strohsnitter W, Kaufman R, Adam E, Noller KL, Herbst AL, Robboy S, Hartge P, Hoover RN 2006 Age at natural menopause in women exposed to diethylstilbestrol in utero. Am J Epidemiol 164:682–688 [PubMed] [Google Scholar]
- Sharara FI, Seifer DB, Flaws JA 1998 Environmental toxicants and female reproduction. Fertil Steril 70:613–622 [PubMed] [Google Scholar]
- Genuis SJ 2006 Health issues and the environment—an emerging paradigm for providers of obstetrical and gynaecological health care. Hum Reprod 21:2201–2208 [PubMed] [Google Scholar]
- Shi Z, Valdez KE, Ting AY, Franczak A, Gum SL, Petroff BK 2007 Ovarian endocrine disruption underlies premature reproductive senescence following environmentally relevant chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biol Reprod 76:198–202 [PubMed] [Google Scholar]
- Chandra A, Martinez GM, Mosher WD, Abma JC, Jones J 2005 Fertility, family planning, and reproductive health of U.S. women: data from the 2002 National Survey of Family Growth. Vital Health Stat 23:1–160 [PubMed] [Google Scholar]
- Kwintkiewicz J, Giudice LC 2009 The interplay of insulin-like growth factors, gonadotropins, and endocrine disruptors in ovarian follicular development and function. Semin Reprod Med 27:43–51 [PubMed] [Google Scholar]
- Minegishi T, Hirakawa T, Abe K, Kishi H, Miyamoto K 2003 Effect of IGF-1 and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the expression of LH receptors during cell differentiation in cultured granulosa cells. Mol Cell Endocrinol 202:123–131 [PubMed] [Google Scholar]
- Holloway AC, Petrik JJ, Younglai EV 2007 Influence of dichlorodiphenylchloroethylene on vascular endothelial growth factor and insulin-like growth factor in human and rat ovarian cells. Reprod Toxicol 24:359–364 [PubMed] [Google Scholar]
- Kwintkiewicz J, Giudice LC, Endocrine disruptor bisphenol A reduces FSH-stimulated cyp19 expression and downstream estradiol production in human granulosa KGN cells. Proc 55th Annual Scientific Meeting of the Society for Gynecologic Investigation, San Diego, CA, 2008 (Abstract) [Google Scholar]
- Kwintkiewicz J, Giudice LC, Endocrine disruptor bisphenol A induces expression of peroxisome proliferators-activated receptor which contributes to down-regulation of FSH-stimulated aromatase expression and estradiol production in human granulosa KGN cells. Proc 41st Annual Meeting of the Society for the Study of Reproduction, Kona, HI, 2008 (Abstract) [Google Scholar]
- Swan SH 2000 Intrauterine exposure to diethylstilbestrol: long-term effects in humans. APMIS 108:793–804 [PubMed] [Google Scholar]
- Jefferies JA, Robboy SJ, O’Brien PC, Bergstralh EJ, Labarthe DR, Barnes AB, Noller KL, Hatab PA, Kaufman RH, Townsend DE 1984 Structural anomalies of the cervix and vagina in women enrolled in the Diethylstilbestrol Adenosis (DESAD) Project. Am J Obstet Gynecol 148:59–66 [PubMed] [Google Scholar]
- Johnson LD, Driscoll SG, Hertig AT, Cole PT, Nickerson RJ 1979 Vaginal adenosis in stillborns and neonates exposed to diethylstilbestrol and steroidal estrogens and progestins. Obstet Gynecol 53:671–679 [PubMed] [Google Scholar]
- Couse JF, Dixon D, Yates M, Moore AB, Ma L, Maas R, Korach KS 2001 Estrogen receptor-α knockout mice exhibit resistance to the developmental effects of neonatal diethylstilbestrol exposure on the female reproductive tract. Dev Biol 238:224–238 [PubMed] [Google Scholar]
- Ma L, Benson GV, Lim H, Dey SK, Maas RL 1998 Abdominal B (AbdB) Hoxa genes: regulation in adult uterus by estrogen and progesterone and repression in müllerian duct by the synthetic estrogen diethylstilbestrol (DES). Dev Biol 197:141–154 [PubMed] [Google Scholar]
- Block K, Kardana A, Igarashi P, Taylor HS 2000 In utero diethylstilbestrol (DES) exposure alters Hox gene expression in the developing müllerian system. FASEB J 14:1101–1108 [PubMed] [Google Scholar]
- Schrager S, Potter BE 2004 Diethylstilbestrol exposure. Am Fam Physician 69:2395–2400 [PubMed] [Google Scholar]
- Buttram Jr VC, Reiter RC 1981 Uterine leiomyomata: etiology, symptomatology, and management. Fertil Steril 36:433–445 [PubMed] [Google Scholar]
- Agency for Healthcare Research and Quality 2007 Management of uterine fibroids: an update of the evidence. Publication no. 07-E011. Bethesda, MD: National Library of Medicine [Google Scholar]
- Wise LA, Palmer JR, Rowlings K, Kaufman RH, Herbst AL, Noller KL, Titus-Ernstoff L, Troisi R, Hatch EE, Robboy SJ 2005 Risk of benign gynecologic tumors in relation to prenatal diethylstilbestrol exposure. Obstet Gynecol 105:167–173 [PubMed] [Google Scholar]
- Baird DD, Newbold R 2005 Prenatal diethylstilbestrol (DES) exposure is associated with uterine leiomyoma development. Reprod Toxicol 20:81–84 [PubMed] [Google Scholar]
- Newbold RR, Moore AB, Dixon D 2002 Characterization of uterine leiomyomas in CD-1 mice following developmental exposure to diethylstilbestrol (DES). Toxicol Pathol 30:611–616 [PubMed] [Google Scholar]
- Everitt JI, Wolf DC, Howe SR, Goldsworthy TL, Walker C 1995 Rodent model of reproductive tract leiomyomata. Clinical and pathological features. Am J Pathol 146:1556–1567 [PMC free article] [PubMed] [Google Scholar]
- Cook JD, Davis BJ, Goewey JA, Berry TD, Walker CL 2007 Identification of a sensitive period for developmental programming that increases risk for uterine leiomyoma in Eker rats. Reprod Sci 14:121–136 [PubMed] [Google Scholar]
- Cook JD, Davis BJ, Cai SL, Barrett JC, Conti CJ, Walker CL 2005 Interaction between genetic susceptibility and early-life environmental exposure determines tumor-suppressor-gene penetrance. Proc Natl Acad Sci USA 102: 8644–8649 [PMC free article] [PubMed] [Google Scholar]
- Bäcklin BM, Eriksson L, Olovsson M 2003 Histology of uterine leiomyoma and occurrence in relation to reproductive activity in the Baltic gray seal (Halichoerus grypus). Vet Pathol 40:175–180 [PubMed] [Google Scholar]
- Simoens S, Hummelshoj L, D’Hooghe T 2007 Endometriosis: cost estimates and methodological perspective. Hum Reprod Update 13:395–404 [PubMed] [Google Scholar]
- Missmer SA, Hankinson SE, Spiegelman D, Barbieri RL, Michels KB, Hunter DJ 2004 In utero exposures and the incidence of endometriosis. Fertil Steril 82:1501–1508 [PubMed] [Google Scholar]
- Rier SE, Martin DC, Bowman RE, Dmowski WP, Becker JL 1993 Endometriosis in rhesus monkeys (Macaca mulatta) following chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam Appl Toxicol 21:433–441 [PubMed] [Google Scholar]
- Rier SE, Turner WE, Martin DC, Morris R, Lucier GW, Clark GC 2001 Serum levels of TCDD and dioxin-like chemicals in Rhesus monkeys chronically exposed to dioxin: correlation of increased serum PCB levels with endometriosis. Toxicol Sci 59:147–159 [PubMed] [Google Scholar]
- Guo SW 2004 The link between exposure to dioxin and endometriosis: a critical reappraisal of primate data. Gynecol Obstet Invest 57:157–173 [PubMed] [Google Scholar]
- Yang JZ, Agarwal SK, Foster WG 2000 Subchronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin modulates the pathophysiology of endometriosis in the cynomolgus monkey. Toxicol Sci 56:374–381 [PubMed] [Google Scholar]
- Cummings AM, Metcalf JL, Birnbaum L 1996 Promotion of endometriosis by 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats and mice: time-dose dependence and species comparison. Toxicol Appl Pharmacol 138:131–139 [PubMed] [Google Scholar]
- Nayyar T, Bruner-Tran KL, Piestrzeniewicz-Ulanska D, Osteen KG 2007 Developmental exposure of mice to TCDD elicits a similar uterine phenotype in adult animals as observed in women with endometriosis. Reprod Toxicol 23:326–336 [PMC free article] [PubMed] [Google Scholar]
- Cobellis L, Latini G, De Felice C, Razzi S, Paris I, Ruggieri F, Mazzeo P, Petraglia F 2003 High plasma concentrations of di-(2-ethylhexyl)-phthalate in women with endometriosis. Hum Reprod 18:1512–1515 [PubMed] [Google Scholar]
- Reddy BS, Rozati R, Reddy BV, Raman NV 2006 Association of phthalate esters with endometriosis in Indian women. BJOG 113:515–520 [PubMed] [Google Scholar]
- Burney R, Giudice LC 2008 Pathogenesis of endometriosis. In: Nezhat CR, ed. Operative gynecologic laparoscopy: principles and techniques. Oxford, UK: Cambridge University Press; 253–259 [Google Scholar]
- Davis DL, Bradlow HL, Wolff M, Woodruff T, Hoel DG, Anton-Culver H 1993 Medical hypothesis: xenoestrogens as preventable causes of breast cancer. Environ Health Perspect 101:372–377 [PMC free article] [PubMed] [Google Scholar]
- Sharpe RM, Skakkebaek NE 1993 Are oestrogens involved in falling sperm counts and disorders of the male reproductive tract? Lancet 341:1392–1395 [PubMed] [Google Scholar]
- Williams EM, Jones L, Vessey MP, McPherson K 1990 Short term increase in risk of breast cancer associated with full term pregnancy. BMJ 300:578–579 [PMC free article] [PubMed] [Google Scholar]
- Lambe M, Hsieh CC, Chan HW, Ekbom A, Trichopoulos D, Adami HO 1996 Parity, age at first and last birth, and risk of breast cancer: a population-based study in Sweden. Breast Cancer Res Treat 38:305–311 [PubMed] [Google Scholar]
- Trichopoulos D 1990 Is breast cancer initiated in utero? Epidemiology 1:95–96 [PubMed] [Google Scholar]
- Hahn WC, Weinberg RA 2002 Modelling the molecular circuitry of cancer. Nat Rev Cancer 2:331–341 [PubMed] [Google Scholar]
- Ho SM, Tang WY, Belmonte de Frausto J, Prins GS 2006 Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res 66:5624–5632 [PMC free article] [PubMed] [Google Scholar]
- Sonnenschein C, Soto AM 1999 The society of cells: cancer and control of cell proliferation. New York: Springer-Verlag. [Google Scholar]
- Sonnenschein C, Soto AM 2008 Theories of carcinogenesis: an emerging perspective. Semin Cancer Biol 18:372–377 [PMC free article] [PubMed] [Google Scholar]
- Soto AM, Sonnenschein C 2004 The somatic mutation theory of cancer: growing problems with the paradigm? Bioessays 26:1097–1107 [PubMed] [Google Scholar]
- Maffini MV, Calabro JM, Soto AM, Sonnenschein C 2005 Stromal regulation of neoplastic development: age-dependent normalization of neoplastic mammary cells by mammary stroma. Am J Pathol 167:1405–1410 [PMC free article] [PubMed] [Google Scholar]
- Markey CM, Rubin BS, Soto AM, Sonnenschein C 2002 Endocrine disruptors: from wingspread to environmental developmental biology. J Steroid Biochem Mol Biol 83:235–244 [PubMed] [Google Scholar]
- Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM 2007 Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology 148:116–127 [PMC free article] [PubMed] [Google Scholar]
- Høyer AP, Grandjean P, Jørgensen T, Brock JW, Hartvig HB 1998 Organochlorine exposure and risk of breast cancer. Lancet 352:1816–1820 [PubMed] [Google Scholar]
- Cohn BA, Wolff MS, Cirillo PM, Sholtz RI 2007 DDT and breast cancer in young women: new data on the significance of age at exposure. Environ Health Perspect 115:1406–1414 [PMC free article] [PubMed] [Google Scholar]
- Ibarluzea Jm J, Fernández MF, Santa-Marina L, Olea- Serrano MF, Rivas AM, Aurrekoetxea JJ, Expósito J, Lorenzo M, Torné P, Villalobos M, Pedraza V, Sasco AJ, Olea N 2004 Breast cancer risk and the combined effect of environmental estrogens. Cancer Causes Control 15: 591–600 [PubMed] [Google Scholar]
- Meyer JS 1977 Cell proliferation in normal human breast ducts, fibroadenomas, and other ductal hyperplasias measured by nuclear labeling with tritiated thymidine. Effects of menstrual phase, age, and oral contraceptive hormones. Hum Pathol 8:67–81 [PubMed] [Google Scholar]
- Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C 2004 Rat mammary gland chemical carcinogenesis: the stroma as a crucial target. J Cell Sci 117:1495–1502 [PubMed] [Google Scholar]
- Palmer JR, Hatch EE, Rosenberg CL, Hartge P, Kaufman RH, Titus-Ernstoff L, Noller KL, Herbst AL, Rao RS, Troisi R, Colton T, Hoover RN 2002 Risk of breast cancer in women exposed to diethylstilbestrol in utero: preliminary results (United States). Cancer Causes Control 13:753–758 [PubMed] [Google Scholar]
- Palmer JR, Wise LA, Hatch EE, Troisi R, Titus-Ernstoff L, Strohsnitter W, Kaufman R, Herbst AL, Noller KL, Hyer M, Hoover RN 2006 Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev 15:1509–1514 [PubMed] [Google Scholar]
- Boylan ES, Calhoon RE 1979 Mammary tumorigenesis in the rat following prenatal exposure to diethylstilbestrol and postnatal treatment with 7,12-dimethylbenz[a]anthracene. J Toxicol Environ Health 5:1059–1071 [PubMed] [Google Scholar]
- Rothschild TC, Boylan ES, Calhoon RE, Vonderhaar BK 1987 Transplacental effects of diethylstilbestrol on mammary development and tumorigenesis in female ACI rats. Cancer Res 47:4508–4516 [PubMed] [Google Scholar]
- Halakivi-Clarke L, Cho E, Onojafe I, Liao DJ, Clarke R 2000 Maternal exposure to tamoxifen during pregnancy increases carcinogen-induced mammary tumorigenesis among female rat offspring. Clin Cancer Res 6:305–308 [PubMed] [Google Scholar]
- Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S 2003 Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 423:545–550 [PubMed] [Google Scholar]
- Brown NM, Manzolillo PA, Zhang JX, Wang J, Lamartiniere CA 1998 Prenatal TCDD and predisposition to mammary cancer in the rat. Carcinogenesis 19:1623–1629 [PubMed] [Google Scholar]
- Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV 2007 Human exposure to bisphenol A (BPA). Reprod Toxicol 24:139–177 [PubMed] [Google Scholar]
- Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL 2008 Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect 116:39–44 [PMC free article] [PubMed] [Google Scholar]
- Richter CA, Birnbaum LS, Farabollini F, Newbold RR, Rubin BS, Talsness CE, Vandenbergh JG, Walser-Kuntz DR, vom Saal FS 2007 In vivo effects of bisphenol A in laboratory rodent studies. Reprod Toxicol 24:199–224 [PMC free article] [PubMed] [Google Scholar]
- Zalko D, Soto AM, Dolo L, Dorio C, Rathahao E, Debrauwer L, Faure R, Cravedi JP 2003 Biotransformations of bisphenol A in a mammalian model: answers and new questions raised by low-dose metabolic fate studies in pregnant CD1 mice. Environ Health Perspect 111:309–319 [PMC free article] [PubMed] [Google Scholar]
- Muñoz-de-Toro M, Markey CM, Wadia PR, Luque EH, Rubin BS, Sonnenschein C, Soto AM 2005 Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice. Endocrinology 146:4138–4147 [PMC free article] [PubMed] [Google Scholar]
- Markey CM, Luque EH, Munoz De Toro M, Sonnenschein C, Soto AM 2001 In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol Reprod 65:1215–1223 [PubMed] [Google Scholar]
- Vandenberg LN, Maffini MV, Schaeberle CM, Ucci AA, Sonnenschein C, Rubin BS, Soto AM 2008 Perinatal exposure to the xenoestrogen bisphenol-A induces mammary intraductal hyperplasias in adult CD-1 mice. Reprod Toxicol 26:210–219 [PMC free article] [PubMed] [Google Scholar]
- Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM 2007 Induction of mammary gland ductal hyperplasia and carcinoma in situ following fetal bisphenol A exposure. Reprod Toxicol 23:383–390 [PMC free article] [PubMed] [Google Scholar]
- Durando M, Kass L, Piva J, Sonnenschein C, Soto AM, Luque EH, Muñoz-de-Toro M 2007 Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect 115:80–86 [PMC free article] [PubMed] [Google Scholar]
- Jenkins S, Raghuraman N, Eltoum I, Carpenter M, Russo J, Lamartiniere CA 7 January 2009 Oral exposure to bisphenol A increases dimethylbenzanthracene-induced mammary cancer in rats. Environ Health Perspect 10.1289/ehp.11751 [PMC free article] [PubMed] [Google Scholar]
- 2005 Third National Report on Human Exposure to Environmental Chemicals. Atlanta: Center for Disease Control and Prevention [Google Scholar]
- Słowikowska-Hilczer J, Szarras-Czapnik M, Kula K 2001 Testicular pathology in 46,XY dysgenetic male pseudohermaphroditism: an approach to pathogenesis of testis cancer. J Androl 22:781–792 [PubMed] [Google Scholar]
- Carlsen E, Giwercman A, Keiding N, Skakkebaek NE 1992 Evidence for decreasing quality of semen during past 50 years. BMJ 305:609–613 [PMC free article] [PubMed] [Google Scholar]
- Auger J, Kunstmann JM, Czyglik F, Jouannet P 1995 Decline in semen quality among fertile men in Paris during the past 20 years. N Engl J Med 332:281–285 [PubMed] [Google Scholar]
- Swan SH, Elkin EP, Fenster L 1997 Have sperm densities declined? A reanalysis of global trend data. Environ Health Perspect 105:1228–1232 [PMC free article] [PubMed] [Google Scholar]
- Bujan L, Mansat A, Pontonnier F, Mieusset R 1996 Time series analysis of sperm concentration in fertile men in Toulouse, France between 1977 and 1992. BMJ 312:471–472 [PMC free article] [PubMed] [Google Scholar]
- Fisch H, Goluboff ET, Olson JH, Feldshuh J, Broder SJ, Barad DH 1996 Semen analyses in 1,283 men from the United States over a 25-year period: no decline in quality. Fertil Steril 65:1009–1014 [PubMed] [Google Scholar]
- Paulsen CA, Berman NG, Wang C 1996 Data from men in greater Seattle area reveals no downward trend in semen quality: further evidence that deterioration of semen quality is not geographically uniform. Fertil Steril 65:1015–1020 [PubMed] [Google Scholar]
- David R, McKee R, Butala J, Barter R, Kaiser M 2001 Esters of aromatic mono-, di-, and tricarboxylic acids, aromatic diacids, and di-, tri-, or polyalcohols. In: Bingham E, Cohrssen B, Powell C, eds. Patty’s toxicology. New York: John Wiley and Sons; 635–932 [Google Scholar]
- 1997 Toxicological profile for di-n-octyl phthalate (DNOP). Atlanta: Agency for Toxic Substances and Disease Registry [Google Scholar]
- 2002 Toxicological profile for di-n-octyl phthalate (DNOP). Atlanta: Agency for Toxic Substances and Disease Registry [Google Scholar]
- 1995 Toxicological profile for di-n-octyl phthalate (DNOP). Atlanta: Agency for Toxic Substances and Disease Registry [Google Scholar]
- 2001 Toxicological profile for di-n-octyl phthalate (DNOP). Atlanta: Agency for Toxic Substances and Disease Registry [Google Scholar]
- Adibi JJ, Perera FP, Jedrychowski W, Camann DE, Barr D, Jacek R, Whyatt RM 2003 Prenatal exposures to phthalates among women in New York City and Krakow, Poland. Environ Health Perspect 111:1719–1722 [PMC free article] [PubMed] [Google Scholar]
- Rudel RA, Camann DE, Spengler JD, Korn LR, Brody JG 2003 Phthalates, alkylphenols, pesticides, polybrominated diphenyl ethers, and other endocrine-disrupting compounds in indoor air and dust. Environ Sci Technol 37:4543–4553 [PubMed] [Google Scholar]
- Green R, Hauser R, Calafat AM, Weuve J, Schettler T, Ringer S, Huttner K, Hu H 2005 Use of di(2-ethylhexyl) phthalate-containing medical products and urinary levels of mono(2-ethylhexyl) phthalate in neonatal intensive care unit infants. Environ Health Perspect 113:1222–1225 [PMC free article] [PubMed] [Google Scholar]
- Duty SM, Silva MJ, Barr DB, Brock JW, Ryan L, Chen Z, Herrick RF, Christiani DC, Hauser R 2003 Phthalate exposure and human semen parameters. Epidemiology 14:269–277 [PubMed] [Google Scholar]
- Hauser R, Meeker JD, Duty S, Silva MJ, Calafat AM 2006 Altered semen quality in relation to urinary concentrations of phthalate monoester and oxidative metabolites. Epidemiology 17:682–691 [PubMed] [Google Scholar]
- Jönsson BA, Richthoff J, Rylander L, Giwercman A, Hagmar L 2005 Urinary phthalate metabolites and biomarkers of reproductive function in young men. Epidemiology 16:487–493 [PubMed] [Google Scholar]
- Longnecker MP, Rogan WJ, Lucier G 1997 The human health effects of DDT (dichlorodiphenyltrichloroethane) and PCBS (polychlorinated biphenyls) and an overview of organochlorines in public health. Annu Rev Public Health 18:211–244 [PubMed] [Google Scholar]
- Brown JF 1994 Determination of PCB metabolic, excretion, and accumulation rates for use as indicators of biological response and relative risk. Environ Sci Technol 28:2295–2305 [PubMed] [Google Scholar]
- Phillips DL, Smith AB, Burse VW, Steele GK, Needham LL, Hannon WH 1989 Half-life of polychlorinated biphenyls in occupationally exposed workers. Arch Environ Health 44:351–354 [PubMed] [Google Scholar]
- Kato Y, Haraguchi K, Shibahara T, Masuda Y, Kimura R 1998 Reduction of thyroid hormone levels by methylsulfonyl metabolites of polychlorinated biphenyl congeners in rats. Arch Toxicol 72:541–544 [PubMed] [Google Scholar]
- Hansen LG 1998 Stepping backward to improve assessment of PCB congener toxicities. Environ Health Perspect 106(Suppl 1):171–189 [PMC free article] [PubMed] [Google Scholar]
- Dallinga JW, Moonen EJ, Dumoulin JC, Evers JL, Geraedts JP, Kleinjans JC 2002 Decreased human semen quality and organochlorine compounds in blood. Hum Reprod 17:1973–1979 [PubMed] [Google Scholar]
- Richthoff J, Rylander L, Jönsson BA, Akesson H, Hagmar L, Nilsson-Ehle P, Stridsberg M, Giwercman A 2003 Serum levels of 2,2′,4,4′,5,5′-hexachlorobiphenyl (CB-153) in relation to markers of reproductive function in young males from the general Swedish population. Environ Health Perspect 111:409–413 [PMC free article] [PubMed] [Google Scholar]
- Hauser R, Chen Z, Pothier L, Ryan L, Altshul L 2003 The relationship between human semen parameters and environmental exposure to polychlorinated biphenyls and p,p’-DDE. Environ Health Perspect 111:1505–1511 [PMC free article] [PubMed] [Google Scholar]
- Rignell-Hydbom A, Rylander L, Giwercman A, Jönsson BA, Nilsson-Ehle P, Hagmar L 2004 Exposure to CB-153 and p,p’-DDE and male reproductive function. Hum Reprod 19:2066–2075 [PubMed] [Google Scholar]
- Guo YL, Hsu PC, Hsu CC, Lambert GH 2000 Semen quality after prenatal exposure to polychlorinated biphenyls and dibenzofurans. Lancet 356:1240–1241 [PubMed] [Google Scholar]
- Hsu PC, Huang W, Yao WJ, Wu MH, Guo YL, Lambert GH 2003 Sperm changes in men exposed to polychlorinated biphenyls and dibenzofurans. JAMA 289:2943–2944 [PubMed] [Google Scholar]
- Mocarelli P, Gerthoux PM, Patterson Jr DG, Milani S, Limonta G, Bertona M, Signorini S, Tramacere P, Colombo L, Crespi C, Brambilla P, Sarto C, Carreri V, Sampson EJ, Turner WE, Needham LL 2008 Dioxin exposure, from infancy through puberty, produces endocrine disruption and affects human semen quality. Environ Health Perspect 116:70–77 [PMC free article] [PubMed] [Google Scholar]
- Abell A, Ernst E, Bonde JP 2000 Semen quality and sexual hormones in greenhouse workers. Scand J Work Environ Health 26:492–500 [PubMed] [Google Scholar]
- Juhler RK, Larsen SB, Meyer O, Jensen ND, Spanò M, Giwercman A, Bonde JP 1999 Human semen quality in relation to dietary pesticide exposure and organic diet. Arch Environ Contam Toxicol 37:415–423 [PubMed] [Google Scholar]
- Oliva A, Spira A, Multigner L 2001 Contribution of environmental factors to the risk of male infertility. Hum Reprod 16:1768–1776 [PubMed] [Google Scholar]
- Larsen SB, Giwercman A, Spanò M, Bonde JP 1998 A longitudinal study of semen quality in pesticide spraying Danish farmers. The ASCLEPIOS Study Group. Reprod Toxicol 12:581–589 [PubMed] [Google Scholar]
- Padungtod C, Savitz DA, Overstreet JW, Christiani DC, Ryan LM, Xu X 2000 Occupational pesticide exposure and semen quality among Chinese workers. J Occup Environ Med 42:982–992 [PubMed] [Google Scholar]
- Kamijima M, Hibi H, Gotoh M, Taki K, Saito I, Wang H, Itohara S, Yamada T, Ichihara G, Shibata E, Nakajima T, Takeuchi Y 2004 A survey of semen indices in insecticide sprayers. J Occup Health 46:109–118 [PubMed] [Google Scholar]
- Lifeng T, Shoulin W, Junmin J, Xuezhao S, Yannan L, Qianli W, Longsheng C 2006 Effects of fenvalerate exposure on semen quality among occupational workers. Contraception 73:92–96 [PubMed] [Google Scholar]
- Whorton MD, Milby TH, Stubbs HA, Avashia BH, Hull EQ 1979 Testicular function among carbaryl-exposed exployees. J Toxicol Environ Health 5:929–941 [PubMed] [Google Scholar]
- Wyrobek AJ, Watchmaker G, Gordon L, Wong K, Moore 2nd D, Whorton D 1981 Sperm shape abnormalities in carbaryl-exposed employees. Environ Health Perspect 40:255–265 [PMC free article] [PubMed] [Google Scholar]
- Tan LF, Sun XZ, Li YN, Ji JM, Wang QL, Chen LS, Bian Q, Wang SL 2005 [Effects of carbaryl production exposure on the sperm and semen quality of occupational male workers]. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 23:87–90 [PubMed] [Google Scholar]
- Swan SH, Kruse RL, Liu F, Barr DB, Drobnis EZ, Redmon JB, Wang C, Brazil C, Overstreet JW 2003 Semen quality in relation to biomarkers of pesticide exposure. Environ Health Perspect 111:1478–1484 [PMC free article] [PubMed] [Google Scholar]
- Meeker JD, Ryan L, Barr DB, Herrick RF, Bennett DH, Bravo R, Hauser R 2004 The relationship of urinary metabolites of carbaryl/naphthalene and chlorpyrifos with human semen quality. Environ Health Perspect 112:1665–1670 [PMC free article] [PubMed] [Google Scholar]
- Boisen KA, Kaleva M, Main KM, Virtanen HE, Haavisto AM, Schmidt IM, Chellakooty M, Damgaard IN, Mau C, Reunanen M, Skakkebaek NE, Toppari J 2004 Difference in prevalence of congenital cryptorchidism in infants between two Nordic countries. Lancet 363:1264–1269 [PubMed] [Google Scholar]
- Paulozzi LJ 1999 International trends in rates of hypospadias and cryptorchidism. Environ Health Perspect 107: 297–302 [PMC free article] [PubMed] [Google Scholar]
- Thonneau PF, Gandia P, Mieusset R 2003 Cryptorchidism: incidence, risk factors, and potential role of environment; an update. J Androl 24:155–162 [PubMed] [Google Scholar]
- Barthold JS, González R 2003 The epidemiology of congenital cryptorchidism, testicular ascent and orchidopexy. J Urol 170:2396–2401 [PubMed] [Google Scholar]
- Dolk H, Vrijheid M, Scott JE, Addor MC, Botting B, de Vigan C, de Walle H, Garne E, Loane M, Pierini A, Garcia-Minaur S, Physick N, Tenconi R, Wiesel A, Calzolari E, Stone D 2004 Toward the effective surveillance of hypospadias. Environ Health Perspect 112:398–402 [PMC free article] [PubMed] [Google Scholar]
- Aho M, Koivisto AM, Tammela T, Auvinen A 2000 Is the incidence of hypospadias increasing? Analysis of Finnish hospital discharge data 1970–1994. Environ Health Perspect 108:463–465 [PMC free article] [PubMed] [Google Scholar]
- Martínez-Frías ML, Prieto D, Prieto L, Bermejo E, Rodríguez-Pinilla E, Cuevas L 2004 Secular decreasing trend of the frequency of hypospadias among newborn male infants in Spain. Birth Defects Res A Clin Mol Teratol 70:75–81 [PubMed] [Google Scholar]
- Longnecker MP, Klebanoff MA, Brock JW, Zhou H, Gray KA, Needham LL, Wilcox AJ 2002 Maternal serum level of 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene and risk of cryptorchidism, hypospadias, and polythelia among male offspring. Am J Epidemiol 155:313–322 [PubMed] [Google Scholar]
- Bhatia R, Shiau R, Petreas M, Weintraub JM, Farhang L, Eskenazi B 2005 Organochlorine pesticides and male genital anomalies in the child health and development studies. Environ Health Perspect 113:220–224 [PMC free article] [PubMed] [Google Scholar]
- Mol NM, Sørensen N, Weihe P, Andersson AM, Jørgensen N, Skakkebaek NE, Keiding N, Grandjean P 2002 Spermaturia and serum hormone concentrations at the age of puberty in boys prenatally exposed to polychlorinated biphenyls. Eur J Endocrinol 146:357–363 [PubMed] [Google Scholar]
- Hosie S, Loff S, Witt K, Niessen K, Waag KL 2000 Is there a correlation between organochlorine compounds and undescended testes? Eur J Pediatr Surg 10:304–309 [PubMed] [Google Scholar]
- Garry VF, Schreinemachers D, Harkins ME, Griffith J 1996 Pesticide appliers, biocides and birth defects in rural Minnesota. Environ Health Perspect 104:394–399 [PMC free article] [PubMed] [Google Scholar]
- García-Rodríguez J, García-Martín M, Nogueras-Ocaña M, de Dios Luna-del-Castillo J, Espigares García M, Olea N, Lardelli-Claret P 1996 Exposure to pesticides and cryptorchidism: geographical evidence of a possible association. Environ Health Perspect 104:1090–1095 [PMC free article] [PubMed] [Google Scholar]
- Kristensen P, Irgens LM, Andersen A, Bye AS, Sundheim L 1997 Birth defects among offspring of Norwegian farmers, 1967–1991. Epidemiology 8:537–544 [PubMed] [Google Scholar]
- Weidner IS, Møller H, Jensen TK, Skakkebaek NE 1998 Cryptorchidism and hypospadias in sons of gardeners and farmers. Environ Health Perspect 106:793–796 [PMC free article] [PubMed] [Google Scholar]
- Pierik FH, Burdorf A, Deddens JA, Juttmann RE, Weber RF 2004 Maternal and paternal risk factors for cryptorchidism and hypospadias: a case-control study in newborn boys. Environ Health Perspect 112:1570–1576 [PMC free article] [PubMed] [Google Scholar]
- Carbone P, Giordano F, Nori F, Mantovani A, Taruscio D, Lauria L, Figà-Talamanca I 2006 Cryptorchidism and hypospadias in the Sicilian district of Ragusa and the use of pesticides. Reprod Toxicol 22:8–12 [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Katsikis I, Piperi C, Kandaraki E, Piouka A, Papavassiliou AG, Panidis D 2008 Increased serum advanced glycation end-products is a distinct finding in lean women with polycystic ovary syndrome (PCOS). Clin Endocrinol (Oxf) 69:634–641 [PubMed] [Google Scholar]
- Adami HO, Bergström R, Möhner M, Zatoñski W, Storm H, Ekbom A, Tretli S, Teppo L, Ziegler H, Rahu M 1994 Testicular cancer in nine northern European countries. Int J Cancer 59:33–38 [PubMed] [Google Scholar]
- 1996 Increase in testicular cancer incidence in six European countries: a birth cohort phenomenon. J Natl Cancer Inst 88:727–733 [PubMed] [Google Scholar]
- Huyghe E, Matsuda T, Thonneau P 2003 Increasing incidence of testicular cancer worldwide: a review. J Urol 170:5–11 [PubMed] [Google Scholar]
- Richiardi L, Bellocco R, Adami HO, Torrång A, Barlow L, Hakulinen T, Rahu M, Stengrevics A, Storm H, Tretli S, Kurtinaitis J, Tyczynski JE, Akre O 2004 Testicular cancer incidence in eight northern European countries: secular and recent trends. Cancer Epidemiol Biomarkers Prev 13:2157–2166 [PubMed] [Google Scholar]
- McGlynn KA, Devesa SS, Sigurdson AJ, Brown LM, Tsao L, Tarone RE 2003 Trends in the incidence of testicular germ cell tumors in the United States. Cancer 97:63–70 [PubMed] [Google Scholar]
- Hemminki K, Li X 2002 Cancer risks in Nordic immigrants and their offspring in Sweden. Eur J Cancer 38:2428–2434 [PubMed] [Google Scholar]
- Henderson BE, Benton B, Jing J, Yu MC, Pike MC 1979 Risk factors for cancer of the testis in young men. Int J Cancer 23:598–602 [PubMed] [Google Scholar]
- Weir HK, Marrett LD, Kreiger N, Darlington GA, Sugar L 2000 Pre-natal and peri-natal exposures and risk of testicular germ-cell cancer. Int J Cancer 87:438–443 [PubMed] [Google Scholar]
- Hardell L, van Bavel B, Lindström G, Carlberg M, Dreifaldt AC, Wijkström H, Starkhammar H, Eriksson M, Hallquist A, Kolmert T 2003 Increased concentrations of polychlorinated biphenyls, hexachlorobenzene, and chlordanes in mothers of men with testicular cancer. Environ Health Perspect 111:930–934 [PMC free article] [PubMed] [Google Scholar]
- Hardell L, Bavel B, Lindström G, Eriksson M, Carlberg M 2006 In utero exposure to persistent organic pollutants in relation to testicular cancer risk. Int J Androl 29:228–234 [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ 2006 Cancer statistics, 2006. CA Cancer J Clin 56:106–130 [PubMed] [Google Scholar]
- Moore R 1947 Endocrinology of neoplastic disease. New York: Oxford University Press; 194 [Google Scholar]
- Huggins C, Hodges CF 1941 Studies on prostatic cancer. I. The effect of castration, of estrogen, and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res 1:293–297 [PubMed] [Google Scholar]
- Leav I, Ho SM, Ofner P, Merk FB, Kwan PW, Damassa D 1988 Biochemical alterations in sex hormone-induced hyperplasia and dysplasia of the dorsolateral prostates of Noble rats. J Natl Cancer Inst 80:1045–1053 [PubMed] [Google Scholar]
- Thomas JA, Keenan EJ 1994 Effects of estrogens on the prostate. J Androl 15:97–99 [PubMed] [Google Scholar]
- Modugno F, Weissfeld JL, Trump DL, Zmuda JM, Shea P, Cauley JA, Ferrell RE 2001 Allelic variants of aromatase and the androgen and estrogen receptors: toward a multigenic model of prostate cancer risk. Clin Cancer Res 7:3092–3096 [PubMed] [Google Scholar]
- Raghow S, Hooshdaran MZ, Katiyar S, Steiner MS 2002 Toremifene prevents prostate cancer in the transgenic adenocarcinoma of mouse prostate model. Cancer Res 62:1370–1376 [PubMed] [Google Scholar]
- Steiner MS, Pound CR 2003 Phase IIA clinical trial to test the efficacy and safety of toremifene in men with high-grade prostatic intraepithelial neoplasia. Clin Prostate Cancer 2:24–31 [PubMed] [Google Scholar]
- Prins GS, Korach KS 2008 The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 73:233–244 [PMC free article] [PubMed] [Google Scholar]
- Henderson BE, Bernstein L, Ross RK, Depue RH, Judd HL 1988 The early in utero oestrogen and testosterone environment of blacks and whites: potential effects on male offspring. Br J Cancer 57:216–218 [PMC free article] [PubMed] [Google Scholar]
- Henderson BE, Ross RK, Pike MC 1991 Toward the primary prevention of cancer. Science 254:1131–1138 [PubMed] [Google Scholar]
- Prins GS, Birch L, Tang WY, Ho SM 2007 Developmental estrogen exposures predispose to prostate carcinogenesis with aging. Reprod Toxicol 23:374–382 [PMC free article] [PubMed] [Google Scholar]
- Morrison H, Savitz D, Semenciw R, Hulka B, Mao Y, Morison D, Wigle D 1993 Farming and prostate cancer mortality. Am J Epidemiol 137:270–280 [PubMed] [Google Scholar]
- Meyer TE, Coker AL, Sanderson M, Symanski E 2007 A case-control study of farming and prostate cancer in African-American and Caucasian men. Occup Environ Med 64:155–160 [PMC free article] [PubMed] [Google Scholar]
- Alavanja MC, Samanic C, Dosemeci M, Lubin J, Tarone R, Lynch CF, Knott C, Thomas K, Hoppin JA, Barker J, Coble J, Sandler DP, Blair A 2003 Use of agricultural pesticides and prostate cancer risk in the Agricultural Health Study cohort. Am J Epidemiol 157:800–814 [PubMed] [Google Scholar]
- Van Maele-Fabry G, Libotte V, Willems J, Lison D 2006 Review and meta-analysis of risk estimates for prostate cancer in pesticide manufacturing workers. Cancer Causes Control 17:353–373 [PubMed] [Google Scholar]
- Mahajan R, Bonner MR, Hoppin JA, Alavanja MC 2006 Phorate exposure and incidence of cancer in the agricultural health study. Environ Health Perspect 114: 1205–1209 [PMC free article] [PubMed] [Google Scholar]
- Usmani KA, Rose RL, Hodgson E 2003 Inhibition and activation of the human liver microsomal and human cytochrome P450 3A4 metabolism of testosterone by deployment-related chemicals. Drug Metab Dispos 31:384–391 [PubMed] [Google Scholar]
- Usmani KA, Cho TM, Rose RL, Hodgson E 2006 Inhibition of the human liver microsomal and human cytochrome P450 1A2 and 3A4 metabolism of estradiol by deployment-related and other chemicals. Drug Metab Dispos 34:1606–1614 [PubMed] [Google Scholar]
- Kester MH, Bulduk S, Tibboel D, Meinl W, Glatt H, Falany CN, Coughtrie MW, Bergman A, Safe SH, Kuiper GG, Schuur AG, Brouwer A, Visser TJ 2000 Potent inhibition of estrogen sulfotransferase by hydroxylated PCB metabolites: a novel pathway explaining the estrogenic activity of PCBs. Endocrinology 141:1897–1900 [PubMed] [Google Scholar]
- Noble RL 1977 The development of prostatic adenocarcinoma in Nb rats following prolonged sex hormone administration. Cancer Res 37:1929–1933 [PubMed] [Google Scholar]
- Driscoll SG, Taylor SH 1980 Effects of prenatal maternal estrogen on the male urogenital system. Obstet Gynecol 56:537–542 [PubMed] [Google Scholar]
- Yonemura CY, Cunha GR, Sugimura Y, Mee SL 1995 Temporal and spatial factors in diethylstilbestrol-induced squamous metaplasia in the developing human prostate. II. Persistent changes after removal of diethylstilbestrol. Acta Anat (Basel) 153:1–11 [PubMed] [Google Scholar]
- Giusti RM, Iwamoto K, Hatch EE 1995 Diethylstilbestrol revisited: a review of the long-term health effects. Ann Intern Med 122:778–788 [PubMed] [Google Scholar]
- Arai Y, Mori T, Suzuki Y, Bern HA 1983 Long-term effects of perinatal exposure to sex steroids and diethylstilbestrol on the reproductive system of male mammals. Int Rev Cytol 84:235–268 [PubMed] [Google Scholar]
- Rajfer J, Coffey DS 1978 Sex steroid imprinting of the immature prostate. Long-term effects. Invest Urol 16:186–190 [PubMed] [Google Scholar]
- Prins GS, Birch L, Habermann H, Chang WY, Tebeau C, Putz O, Bieberich C 2001 Influence of neonatal estrogens on rat prostate development. Reprod Fertil Dev 13:241–252 [PubMed] [Google Scholar]
- Huang L, Pu Y, Alam S, Birch L, Prins GS 2004 Estrogenic regulation of signaling pathways and homeobox genes during rat prostate development. J Androl 25:330–337 [PubMed] [Google Scholar]
- Gupta C 2000 The role of estrogen receptor, androgen receptor and growth factors in diethylstilbestrol-induced programming of prostate differentiation. Urol Res 28:223–229 [PubMed] [Google Scholar]
- Lemmen JG, Arends RJ, van der Saag PT, van der Burg B 2004 In vivo imaging of activated estrogen receptors in utero by estrogens and bisphenol A. Environ Health Perspect 112:1544–1549 [PMC free article] [PubMed] [Google Scholar]
- Kurosawa T, Hiroi H, Tsutsumi O, Ishikawa T, Osuga Y, Fujiwara T, Inoue S, Muramatsu M, Momoeda M, Taketani Y 2002 The activity of bisphenol A depends on both the estrogen receptor subtype and the cell type. Endocr J 49:465–471 [PubMed] [Google Scholar]
- Song KH, Lee K, Choi HS 2002 Endocrine disruptor bisphenol A induces orphan nuclear receptor Nur77 gene expression and steroidogenesis in mouse testicular Leydig cells. Endocrinology 143:2208–2215 [PubMed] [Google Scholar]
- Walsh DE, Dockery P, Doolan CM 2005 Estrogen receptor independent rapid non-genomic effects of environmental estrogens on [Ca2+]i in human breast cancer cells. Mol Cell Endocrinol 230:23–30 [PubMed] [Google Scholar]
- Keri RA, Ho SM, Hunt PA, Knudsen KE, Soto AM, Prins GS 2007 An evaluation of evidence for the carcinogenic activity of bisphenol A. Reprod Toxicol 24:240–252 [PMC free article] [PubMed] [Google Scholar]
- Wetherill YB, Fisher NL, Staubach A, Danielsen M, de Vere White RW, Knudsen KE 2005 Xenoestrogen action in prostate cancer: pleiotropic effects dependent on androgen receptor status. Cancer Res 65:54–65 [PubMed] [Google Scholar]
- Wetherill YB, Hess-Wilson JK, Comstock CE, Shah SA, Buncher CR, Sallans L, Limbach PA, Schwemberger S, Babcock GF, Knudsen KE 2006 Bisphenol A facilitates bypass of androgen ablation therapy in prostate cancer. Mol Cancer Ther 5:3181–3190 [PubMed] [Google Scholar]
- Prins GS, Tang WY, Belmonte J, Ho SM 2008 Perinatal exposure to oestradiol and bisphenol A alters the prostate epigenome and increases susceptibility to carcinogenesis. Basic Clin Pharmacol Toxicol 102:134–138 [PMC free article] [PubMed] [Google Scholar]
- Hardell L, Andersson SO, Carlberg M, Bohr L, van Bavel B, Lindström G, Björnfoth H, Ginman C 2006 Adipose tissue concentrations of persistent organic pollutants and the risk of prostate cancer. J Occup Environ Med 48:700–707 [PubMed] [Google Scholar]
- Prince MM, Ruder AM, Hein MJ, Waters MA, Whelan EA, Nilsen N, Ward EM, Schnorr TM, Laber PA, Davis-King KE 2006 Mortality and exposure response among 14,458 electrical capacitor manufacturing workers exposed to polychlorinated biphenyls (PCBs). Environ Health Perspect 114:1508–1514 [PMC free article] [PubMed] [Google Scholar]
- Ritchie JM, Vial SL, Fuortes LJ, Guo H, Reedy VE, Smith EM 2003 Organochlorines and risk of prostate cancer. J Occup Environ Med 45:692–702 [PubMed] [Google Scholar]
- Charles LE, Loomis D, Shy CM, Newman B, Millikan R, Nylander-French LA, Couper D 2003 Electromagnetic fields, polychlorinated biphenyls, and prostate cancer mortality in electric utility workers. Am J Epidemiol 157:683–691 [PubMed] [Google Scholar]
- Schlumpf M, Schmid P, Durrer S, Conscience M, Maerkel K, Henseler M, Gruetter M, Herzog I, Reolon S, Ceccatelli R, Faass O, Stutz E, Jarry H, Wuttke W, Lichtensteiger W 2004 Endocrine activity and developmental toxicity of cosmetic UV filters–an update. Toxicology 205:113–122 [PubMed] [Google Scholar]
- Schlumpf M, Jarry H, Wuttke W, Ma R, Lichtensteiger W 2004 Estrogenic activity and estrogen receptor β binding of the UV filter 3-benzylidene camphor. Comparison with 4-methylbenzylidene camphor. Toxicology 199:109–120 [PubMed] [Google Scholar]
- Hofkamp L, Bradley S, Tresguerres J, Lichtensteiger W, Schlumpf M, Timms B 2008 Region-specific growth effects in the developing rat prostate following fetal exposure to estrogenic ultraviolet filters. Environ Health Perspect 116:867–872 [PMC free article] [PubMed] [Google Scholar]
- Parent ME, Siemiatycki J 2001 Occupation and prostate cancer. Epidemiol Rev 23:138–143 [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Liu J, Webber MM, Waalkes MP 2007 Estrogen signaling and disruption of androgen metabolism in acquired androgen-independence during cadmium carcinogenesis in human prostate epithelial cells. Prostate 67:135–145 [PubMed] [Google Scholar]
- Waalkes MP 2000 Cadmium carcinogenesis in review. J Inorg Biochem 79:241–244 [PubMed] [Google Scholar]
- Watson WH, Yager JD 2007 Arsenic: extension of its endocrine disruption potential to interference with estrogen receptor-mediated signaling. Toxicol Sci 98:1–4 [PubMed] [Google Scholar]
- Davey JC, Bodwell JE, Gosse JA, Hamilton JW 2007 Arsenic as an endocrine disruptor: effects of arsenic on estrogen receptor-mediated gene expression in vivo and in cell culture. Toxicol Sci 98:75–86 [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Webber MM, Waalkes MP 2007 Mechanisms of acquired androgen independence during arsenic-induced malignant transformation of human prostate epithelial cells. Environ Health Perspect 115:243–247 [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Kuo TL, Wu MM 1988 Arsenic and cancers. Lancet 1:414–415 [PubMed] [Google Scholar]
- Lewis DR, Southwick JW, Ouellet-Hellstrom R, Rench J, Calderon RL 1999 Drinking water arsenic in Utah: a cohort mortality study. Environ Health Perspect 107:359–365 [PMC free article] [PubMed] [Google Scholar]
- Kavlock R, Cummings A 2005 Mode of action: inhibition of androgen receptor function—vinclozolin-induced malformations in reproductive development. Crit Rev Toxicol 35:721–726 [PubMed] [Google Scholar]
- Yu WJ, Lee BJ, Nam SY, Ahn B, Hong JT, Do JC, Kim YC, Lee YS, Yun YW 2004 Reproductive disorders in pubertal and adult phase of the male rats exposed to vinclozolin during puberty. J Vet Med Sci 66:847–853 [PubMed] [Google Scholar]
- Cowin PA, Foster P, Pedersen J, Hedwards S, McPherson SJ, Risbridger GP 2008 Early-onset endocrine disruptor-induced prostatitis in the rat. Environ Health Perspect 116:923–929 [PMC free article] [PubMed] [Google Scholar]
- Cocco P, Benichou J 1998 Mortality from cancer of the male reproductive tract and environmental exposure to the anti-androgen p,p’-dichlorodiphenyldichloroethylene in the United States. Oncology 55:334–339 [PubMed] [Google Scholar]
- Gore A 2007 Endocrine-disrupting chemicals: from basic research to clinical practice. In: Conn PM, ed. Contemporary endocrinology. Totowa, NJ: Humana Press [Google Scholar]
- Walker DM, Gore AC 2007 Endocrine-disrupting chemicals and the brain. In: Gore AC, ed. Endocrine-disrupting chemicals: from basic research to clinical practice. Totowa, NJ: Humana Press; 63–109 [Google Scholar]
- Gore AC 2008 Neuroendocrine systems as targets for environmental endocrine-disrupting chemicals. Fertil Steril 89:e101–e102 [PMC free article] [PubMed] [Google Scholar]
- Gore A 2002 GnRH: the master molecule of reproduction. Norwell, MA: Kluwer Academic Publishers [Google Scholar]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Gröne HJ, Todman MG, Korach KS, Greiner E, Pérez CA, Schütz G, Herbison AE 2006 Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 52:271–280 [PMC free article] [PubMed] [Google Scholar]
- Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI 1990 Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5:1–10 [PubMed] [Google Scholar]
- Gore AC, Wu TJ, Oung T, Lee JB, Woller MJ 2002 A novel mechanism for endocrine-disrupting effects of polychlorinated biphenyls: direct effects on gonadotropin-releasing hormone neurones. J Neuroendocrinol 14:814–823 [PubMed] [Google Scholar]
- Gore AC 2002 Organochlorine pesticides directly regulate gonadotropin-releasing hormone gene expression and biosynthesis in the GT1–7 hypothalamic cell line. Mol Cell Endocrinol 192:157–170 [PubMed] [Google Scholar]
- Cook R, Calabrese EJ 2006 The importance of hormesis to public health. Environ Health Perspect 114:1631–1635 [PMC free article] [PubMed] [Google Scholar]
- Gore AC, Heindel JJ, Zoeller RT 2006 Endocrine disruption for endocrinologists (and others). Endocrinology 147:S1–S3 [PubMed] [Google Scholar]
- Rasier G, Parent AS, Gérard A, Denooz R, Lebrethon MC, Charlier C, Bourguignon JP 2008 Mechanisms of interaction of endocrine-disrupting chemicals with glutamate-evoked secretion of gonadotropin-releasing hormone. Toxicol Sci 102:33–41 [PubMed] [Google Scholar]
- McGarvey C, Cates PA, Brooks A, Swanson IA, Milligan SR, Coen CW, O’Byrne KT 2001 Phytoestrogens and gonadotropin-releasing hormone pulse generator activity and pituitary luteinizing hormone release in the rat. Endocrinology 142:1202–1208 [PubMed] [Google Scholar]
- Patisaul HB, Fortino AE, Polston EK 2007 Differential disruption of nuclear volume and neuronal phenotype in the preoptic area by neonatal exposure to genistein and bisphenol-A. Neurotoxicology 28:1–12 [PubMed] [Google Scholar]
- Gore AC 2001 Environmental toxicant effects on neuroendocrine function. Endocrine 14:235–246 [PubMed] [Google Scholar]
- Bisenius ES, Veeramachaneni DN, Sammonds GE, Tobet S 2006 Sex differences and the development of the rabbit brain: effects of vinclozolin. Biol Reprod 75:469–476 [PubMed] [Google Scholar]
- Khan IA, Thomas P 2001 Disruption of neuroendocrine control of luteinizing hormone secretion by aroclor 1254 involves inhibition of hypothalamic tryptophan hydroxylase activity. Biol Reprod 64:955–964 [PubMed] [Google Scholar]
- Gore AC 2008 Developmental programming and endocrine disruptor effects on reproductive neuroendocrinology. Front Neuroendocrinol 29:358–374 [PMC free article] [PubMed] [Google Scholar]
- George FW, Wilson JD 1978 Conversion of androgen to estrogen by the human fetal ovary. J Clin Endocrinol Metab 47:550–555 [PubMed] [Google Scholar]
- Steinberg RM, Juenger TE, Gore AC 2007 The effects of prenatal PCBs on adult female paced mating reproductive behaviors in rats. Horm Behav 51:364–372 [PMC free article] [PubMed] [Google Scholar]
- Chung YW, Clemens LG 1999 Effects of perinatal exposure to polychlorinated biphenyls on development of female sexual behavior. Bull Environ Contam Toxicol 62:664–670 [PubMed] [Google Scholar]
- Chung YW, Nunez AA, Clemens LG 2001 Effects of neonatal polychlorinated biphenyl exposure on female sexual behavior. Physiol Behav 74:363–370 [PubMed] [Google Scholar]
- Patisaul HB, Luskin JR, Wilson ME 2004 A soy supplement and tamoxifen inhibit sexual behavior in female rats. Horm Behav 45:270–277 [PubMed] [Google Scholar]
- Whitten PL, Lewis C, Russell E, Naftolin F 1995 Phytoestrogen influences on the development of behavior and gonadotropin function. Proc Soc Exp Biol Med 208:82–86 [PubMed] [Google Scholar]
- Kouki T, Okamoto M, Wada S, Kishitake M, Yamanouchi K 2005 Suppressive effect of neonatal treatment with a phytoestrogen, coumestrol, on lordosis and estrous cycle in female rats. Brain Res Bull 64:449–454 [PubMed] [Google Scholar]
- Crews D, Gore AC, Hsu TS, Dangleben NL, Spinetta M, Schallert T, Anway MD, Skinner MK 2007 Transgenerational epigenetic imprints on mate preference. Proc Natl Acad Sci USA 104:5942–5946 [PMC free article] [PubMed] [Google Scholar]
- Harvey PW, Everett DJ, Springall CJ 2007 Adrenal toxicology: a strategy for assessment of functional toxicity to the adrenal cortex and steroidogenesis. J Appl Toxicol 27:103–115 [PubMed] [Google Scholar]
- Zoeller RT, Tan SW, Tyl RW 2007 General background on the hypothalamic-pituitary-thyroid (HPT) axis. Crit Rev Toxicol 37:11–53 [PubMed] [Google Scholar]
- Khan MA, Hansen LG 2003 Ortho-substituted polychlorinated biphenyl (PCB) congeners (95 or 101) decrease pituitary response to thyrotropin releasing hormone. Toxicol Lett 144:173–182 [PubMed] [Google Scholar]
- Kuriyama SN, Wanner A, Fidalgo-Neto AA, Talsness CE, Koerner W, Chahoud I 2007 Developmental exposure to low-dose PBDE-99: tissue distribution and thyroid hormone levels. Toxicology 242:80–90 [PubMed] [Google Scholar]
- Sørmo EG, Jüssi I, Jüssi M, Braathen M, Skaare JU, Jenssen BM 2005 Thyroid hormone status in gray seal (Halichoerus grypus) pups from the Baltic Sea and the Atlantic Ocean in relation to organochlorine pollutants. Environ Toxicol Chem 24:610–616 [PubMed] [Google Scholar]
- Newbold RR, Padilla-Banks E, Jefferson WN 2006 Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology 147:S11–S17 [PubMed] [Google Scholar]
- Noaksson E, Tjärnlund U, Bosveld AT, Balk L 2001 Evidence for endocrine disruption in perch (Perca fluviatilis) and roach (Rutilus rutilus) in a remote Swedish lake in the vicinity of a public refuse dump. Toxicol Appl Pharmacol 174:160–176 [PubMed] [Google Scholar]
- Simerly RB, Chang C, Muramatsu M, Swanson LW 1990 Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol 294:76–95 [PubMed] [Google Scholar]
- Quadros PS, Pfau JL, Goldstein AY, De Vries GJ, Wagner CK 2002 Sex differences in progesterone receptor expression: a potential mechanism for estradiol-mediated sexual differentiation. Endocrinology 143:3727–3739 [PubMed] [Google Scholar]
- Chakraborty TR, Gore AC 2004 Aging-related changes in ovarian hormones, their receptors, and neuroendocrine function. Exp Biol Med (Maywood) 229:977–987 [PubMed] [Google Scholar]
- Tokumoto T, Tokumoto M, Thomas P 2007 Interactions of diethylstilbestrol (DES) and DES analogs with membrane progestin receptor-α and the correlation with their nongenomic progestin activities. Endocrinology 148: 3459–3467 [PubMed] [Google Scholar]
- Thomas P, Doughty K 2004 Disruption of rapid, nongenomic steroid actions by environmental chemicals: interference with progestin stimulation of sperm motility in Atlantic croaker. Environ Sci Technol 38:6328–6332 [PubMed] [Google Scholar]
- Watson CS, Bulayeva NN, Wozniak AL, Alyea RA 2007 Xenoestrogens are potent activators of nongenomic estrogenic responses. Steroids 72:124–134 [PMC free article] [PubMed] [Google Scholar]
- Brucker-Davis F 1998 Effects of environmental synthetic chemicals on thyroid function. Thyroid 8:827–856 [PubMed] [Google Scholar]
- Howdeshell KL 2002 A model of the development of the brain as a construct of the thyroid system. Environ Health Perspect 110(Suppl 3):337–348 [PMC free article] [PubMed] [Google Scholar]
- Delange F 1989 Cassava and the thyroid. In: Gaitan E, ed. Environmental goitrogenesis. Boca Raton, FL: CRC Press, Inc.; 173–194 [Google Scholar]
- Gaitan E 1989 Environmental goitrogenesis. Boca Raton, FL: CRC Press, Inc. [Google Scholar]
- Carrasco N 2000 Thyroid iodide transport: the Na+/I− symporter (NIS). In: Braverman LE, Utiger RD, eds. Werner & Ingbar’s The Thyroid: a fundamental and clinical text. 8 th ed. Philadelphia: Lippincott, Williams & Wilkins; 52–61 [Google Scholar]
- Zimmermann MB 2007 The adverse effects of mild-to-moderate iodine deficiency during pregnancy and childhood: a review. Thyroid 17:829–835 [PubMed] [Google Scholar]
- National Research Council 2005 Health implications of perchlorate ingestion. Washington, DC: National Academies Press [Google Scholar]
- Murray CW, Egan SK, Kim H, Beru N, Bolger PM 2008 US Food and Drug Administration’s Total Diet Study: dietary intake of perchlorate and iodine. J Expo Sci Environ Epidemiol 18:571–580 [PubMed] [Google Scholar]
- Blount BC, Valentin-Blasini L, Osterloh JD, Mauldin JP, Pirkle JL 2007 Perchlorate exposure of the US population, 2001–2002. J Expo Sci Environ Epidemiol 17:400–407 [PubMed] [Google Scholar]
- Greer MA, Goodman G, Pleus RC, Greer SE 2002 Health effects assessment for environmental perchlorate contamination: the dose response for inhibition of thyroidal radioiodine uptake in humans. Environ Health Perspect 110:927–937 [PMC free article] [PubMed] [Google Scholar]
- Blount BC, Pirkle JL, Osterloh JD, Valentin-Blasini L, Caldwell KL 2006 Urinary perchlorate and thyroid hormone levels in adolescent and adult men and women living in the United States. Environ Health Perspect 114:1865–1871 [PMC free article] [PubMed] [Google Scholar]
- Steinmaus C, Miller MD, Howd R 2007 Impact of smoking and thiocyanate on perchlorate and thyroid hormone associations in the 2001–2002 national health and nutrition examination survey. Environ Health Perspect 115:1333–1338 [PMC free article] [PubMed] [Google Scholar]
- Zoeller RT, Rovet J 2004 Timing of thyroid hormone action in the developing brain: clinical observations and experimental findings. J Neuroendocrinol 16:809–818 [PubMed] [Google Scholar]
- Pearce EN, Leung AM, Blount BC, Bazrafshan HR, He X, Pino S, Valentin-Blasini L, Braverman LE 2007 Breast milk iodine and perchlorate concentrations in lactating Boston-area women. J Clin Endocrinol Metab 92:1673–1677 [PubMed] [Google Scholar]
- Ginsberg GL, Hattis DB, Zoeller RT, Rice DC 2007 Evaluation of the U.S. EPA/OSWER preliminary remediation goal for perchlorate in groundwater: focus on exposure to nursing infants. Environ Health Perspect 115:361–369 [PMC free article] [PubMed] [Google Scholar]
- Amitai Y, Winston G, Sack J, Wasser J, Lewis M, Blount BC, Valentin-Blasini L, Fisher N, Israeli A, Leventhal A 2007 Gestational exposure to high perchlorate concentrations in drinking water and neonatal thyroxine levels. Thyroid 17:843–850 [PubMed] [Google Scholar]
- Wolff J 1998 Perchlorate and the thyroid gland. Pharmacol Rev 50:89–105 [PubMed] [Google Scholar]
- De Groef B, Decallonne BR, Van der Geyten S, Darras VM, Bouillon R 2006 Perchlorate versus other environmental sodium/iodide symporter inhibitors: potential thyroid-related health effects. Eur J Endocrinol 155:17–25 [PubMed] [Google Scholar]
- Taurog A 2000 Hormone synthesis: thyroid iodine metabolism. In: Braverman L, Utiger R, eds. Werner & Ingbar’s The Thyroid: a fundamental and clinical text. 8th ed. Philadelphia: Lippincott, Williams & Wilkins; 61–85 [Google Scholar]
- Taurog A 1964 The biosynthesis of thyroxine. Mayo Clin Proc 39:569–585 [PubMed] [Google Scholar]
- Cooper DS 2003 Antithyroid drugs in the management of patients with Graves’ disease: an evidence-based approach to therapeutic controversies. J Clin Endocrinol Metab 88:3474–3481 [PubMed] [Google Scholar]
- Mestman JH 1998 Hyperthyroidism in pregnancy. Endocrinol Metab Clin North Am 27:127–149 [PubMed] [Google Scholar]
- Engler H, Taurog A, Nakashima T 1982 Mechanism of inactivation of thyroid peroxidase by thioureylene drugs. Biochem Pharmacol 31:3801–3806 [PubMed] [Google Scholar]
- Doerge DR, Sheehan DM 2002 Goitrogenic and estrogenic activity of soy isoflavones. Environ Health Perspect 110(Suppl 3):349–353 [PMC free article] [PubMed] [Google Scholar]
- Labib M, Gama R, Wright J, Marks V, Robins D 1989 Dietary maladvice as a cause of hypothyroidism and short stature. Brit Med J 298:232–233 [PMC free article] [PubMed] [Google Scholar]
- Chorazy PA, Himelhoch S, Hopwood NJ, Greger NG, Postellon DC 1995 Persistent hypothyroidism in an infant receiving a soy formula: case report and review of the literature. Pediatrics 96:148–150 [PubMed] [Google Scholar]
- Jabbar MA, Larrea J, Shaw RA 1997 Abnormal thyroid function tests in infants with congenital hypothyroidism: the influence of soy-based formula. J Am Coll Nutr 16:280–282 [PubMed] [Google Scholar]
- Fort P, Moses N, Fasano M, Goldberg T, Lifshitz F 1990 Breast and soy-formula feedings in early infancy and the prevalence of autoimmune thyroid disease in children. J Am Coll Nutr 9:164–167 [PubMed] [Google Scholar]
- Boker LK, Van der Schouw YT, De Kleijn MJ, Jacques PF, Grobbee DE, Peeters PH 2002 Intake of dietary phytoestrogens by Dutch women. J Nutr 132:1319–1328 [PubMed] [Google Scholar]
- Schussler GC 2000 The thyroxine-binding proteins. Thyroid 10:141–149 [PubMed] [Google Scholar]
- Vranckx R, Savu L, Maya M, Nunez EA 1990 Characterization of a major development-regulated serum thyroxine-binding globulin in the euthyroid mouse. Biochem J 271:373–379 [PMC free article] [PubMed] [Google Scholar]
- Robbins J 2000 Thyroid hormone transport proteins and the physiology of hormone binding. In: Braverman L, Utiger R, eds. Werner & Ingbar’s The Thyroid: a fundamental and clinical text. 8th ed. Philadelphia: Lippincott, Williams and Wilkins; 105–120 [Google Scholar]
- Refetoff S 1989 Inherited thyroxine-binding globulin abnormalities in man. Endocr Rev 10:275–293 [PubMed] [Google Scholar]
- Mendel CM, Weisiger RA, Jones AL, Cavalieri RR 1987 Thyroid hormone-binding proteins in plasma facilitate uniform distribution of thyroxine within tissues: a perfused rat liver study. Endocrinology 120:1742–1749 [PubMed] [Google Scholar]
- Power DM, Elias NP, Richardson SJ, Mendes J, Soares CM, Santos CR 2000 Evolution of the thyroid hormone-binding protein, transthyretin. Gen Comp Endocrinol 119:241–255 [PubMed] [Google Scholar]
- Zheng W, Lu YM, Lu GY, Zhao Q, Cheung O, Blaner WS 2001 Transthyretin, thyroxine, and retinol-binding protein in human cerebrospinal fluid: effect of lead exposure. Toxicol Sci 61:107–114 [PMC free article] [PubMed] [Google Scholar]
- Robbins J 2002 Transthyretin from discovery to now. Clin Chem Lab Med 40:1183–1190 [PubMed] [Google Scholar]
- Palha JA, Nissanov J, Fernandes R, Sousa JC, Bertrand L, Dratman MB, Morreale de Escobar G, Gottesman M, Saraiva MJ 2002 Thyroid hormone distribution in the mouse brain: the role of transthyretin. Neuroscience 113:837–847 [PubMed] [Google Scholar]
- Palha JA, Fernandes R, de Escobar GM, Episkopou V, Gottesman M, Saraiva MJ 2000 Transthyretin regulates thyroid hormone levels in the choroid plexus, but not in the brain parenchyma: study in a transthyretin-null mouse model. Endocrinology 141:3267–3272 [PubMed] [Google Scholar]
- Meerts IA, van Zanden JJ, Luijks EA, van Leeuwen-Bol I, Marsh G, Jakobsson E, Bergman A, Brouwer A 2000 Potent competitive interactions of some brominated flame retardants and related compounds with human transthyretin in vitro. Toxicol Sci 56:95–104 [PubMed] [Google Scholar]
- Chauhan KR, Kodavanti PR, McKinney JD 2000 Assessing the role of ortho-substitution on polychlorinated biphenyl binding to transthyretin, a thyroxine transport protein. Toxicol Appl Pharmacol 162:10–21 [PubMed] [Google Scholar]
- Lans MC, Klasson-Wehler E, Willemsen M, Meussen E, Safe S, Brouwer A 1993 Structure-dependent, competitive interaction of hydroxy-polychlorobiphenyls, -dibenzo-p-dioxins, and -dibenzofurans with human transthyretin. Chem Biol Interact 88:7–21 [PubMed] [Google Scholar]
- Brouwer A, Morse DC, Lans MC, Schuur AG, Murk AJ, Klasson-Wehler E, Bergman A, Visser TJ 1998 Interactions of persistent environmental organohalogens with the thyroid hormone system: mechanisms and possible consequences for animal and human health. Toxicol Ind Health 14:59–84 [PubMed] [Google Scholar]
- Richardson SJ, Lemkine GF, Alfama G, Hassani Z, Demeneix BA 2007 Cell division and apoptosis in the adult neural stem cell niche are differentially affected in transthyretin null mice. Neurosci Lett 421:234–238 [PubMed] [Google Scholar]
- Meerts IA, Assink Y, Cenijn PH, Van Den Berg JH, Weijers BM, Bergman A, Koeman JH, Brouwer A 2002 Placental transfer of a hydroxylated polychlorinated biphenyl and effects on fetal and maternal thyroid hormone homeostasis in the rat. Toxicol Sci 68:361–371 [PubMed] [Google Scholar]
- Fisher JW, Campbell J, Muralidhara S, Bruckner JV, Ferguson D, Mumtaz M, Harmon B, Hedge JM, Crofton KM, Kim H, Almekinder TL 2006 Effect of PCB 126 on hepatic metabolism of thyroxine and perturbations in the hypothalamic-pituitary-thyroid axis in the rat. Toxicol Sci 90:87–95 [PubMed] [Google Scholar]
- Glatt CM, Ouyang M, Welsh W, Green JW, Connor JO, Frame SR, Everds NE, Poindexter G, Snajdr S, Delker DA 2005 Molecular characterization of thyroid toxicity: anchoring gene expression profiles to biochemical and pathologic end points. Environ Health Perspect 113:1354–1361 [PMC free article] [PubMed] [Google Scholar]
- Tseng LH, Li MH, Tsai SS, Lee CW, Pan MH, Yao WJ, Hsu PC 2008 Developmental exposure to decabromodiphenyl ether (PBDE 209): effects on thyroid hormone and hepatic enzyme activity in male mouse offspring. Chemosphere 70:640–647 [PubMed] [Google Scholar]
- Kretschmer XC, Baldwin WS 2005 CAR and PXR: xenosensors of endocrine disrupters? Chem Biol Interact 155:111–128 [PubMed] [Google Scholar]
- Rathore M, Bhatnagar P, Mathur D, Saxena GN 2002 Burden of organochlorine pesticides in blood and its effect on thyroid hormones in women. Sci Total Environ 295:207–215 [PubMed] [Google Scholar]
- Langer P 2008 Persistent organochlorinated pollutants (PCB, DDE, HCB, dioxins, furans) and the thyroid—review 2008. Endocr Regul 42:79–104 [PubMed] [Google Scholar]
- Köhrle J 2007 Thyroid hormone transporters in health and disease: advances in thyroid hormone deiodination. Best Pract Res Clin Endocrinol Metab 21:173–191 [PubMed] [Google Scholar]
- Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE 2005 Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet 77:41–53 [PMC free article] [PubMed] [Google Scholar]
- St Germain DL, Hernandez A, Schneider MJ, Galton VA 2005 Insights into the role of deiodinases from studies of genetically modified animals. Thyroid 15:905–916 [PubMed] [Google Scholar]
- Schneider MJ, Fiering SN, Pallud SE, Parlow AF, St Germain DL, Galton VA 2001 Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol 15:2137–2148 [PubMed] [Google Scholar]
- Kato Y, Ikushiro S, Haraguchi K, Yamazaki T, Ito Y, Suzuki H, Kimura R, Yamada S, Inoue T, Degawa M 2004 A possible mechanism for decrease in serum thyroxine level by polychlorinated biphenyls in Wistar and Gunn rats. Toxicol Sci 81:309–315 [PubMed] [Google Scholar]
- Morse DC, Wehler EK, Wesseling W, Koeman JH, Brouwer A 1996 Alterations in rat brain thyroid hormone status following pre- and postnatal exposure to polychlorinated biphenyls (Aroclor 1254). Toxicol Appl Pharmacol 136:269–279 [PubMed] [Google Scholar]
- McKinney JD, Waller CL 1994 Polychlorinated biphenyls as hormonally active structural analogues. Environ Health Perspect 102:290–297 [PMC free article] [PubMed] [Google Scholar]
- Chana A, Concejero MA, de Frutos M, González MJ, Herradón B 2002 Computational studies on biphenyl derivatives. Analysis of the conformational mobility, molecular electrostatic potential, and dipole moment of chlorinated biphenyl: searching for the rationalization of the selective toxicity of polychlorinated biphenyls (PCBs). Chem Res Toxicol 15:1514–1526 [PubMed] [Google Scholar]
- Breivik K, Sweetman A, Pacyna JM, Jones KC 2002 Towards a global historical emission inventory for selected PCB congeners—a mass balance approach. 1. Global production and consumption. Sci Total Environ 290:181–198 [PubMed] [Google Scholar]
- Fisher BE1999 Most unwanted. Environ Health Perspect 107:A18–A23 [PMC free article] [PubMed] [Google Scholar]
- Huisman M, Koopman-Esseboom C, Lanting CI, van der Paauw CG, Tuinstra LG, Fidler V, Weisglas-Kuperus N, Sauer PJ, Boersma ER, Touwen BC 1995 Neurological condition in 18-month-old children perinatally exposed to polychlorinated biphenyls and dioxins. Early Hum Dev 43:165–176 [PubMed] [Google Scholar]
- Jackson TA, Richer JK, Bain DL, Takimoto GS, Tung L, Horwitz KB 1997 The partial agonist activity of antagonist-occupied steroid receptors is controlled by a novel hinge domain-binding coactivator L7/SPA and the corepressors N-CoR or SMRT. Mol Endocrinol 11:693–705 [PubMed] [Google Scholar]
- Osius N, Karmaus W, Kruse H, Witten J 1999 Exposure to polychlorinated biphenyls and levels of thyroid hormones in children. Environ Health Perspect 107:843–849 [PMC free article] [PubMed] [Google Scholar]
- Ayotte P, Muckle G, Jacobson JL, Jacobson SW, Dewailly E 2003 Assessment of pre- and postnatal exposure to polychlorinated biphenyls: lessons from the Inuit Cohort Study. Environ Health Perspect 111:1253–1258 [PMC free article] [PubMed] [Google Scholar]
- Walkowiak J, Wiener JA, Fastabend A, Heinzow B, Krämer U, Schmidt E, Steingrüber HJ, Wundram S, Winneke G 2001 Environmental exposure to polychlorinated biphenyls and quality of the home environment: effects on psychodevelopment in early childhood. Lancet 358:1602–1607 [PubMed] [Google Scholar]
- Zoeller RT, Dowling AL, Vas AA 2000 Developmental exposure to polychlorinated biphenyls exerts thyroid hormone-like effects on the expression of RC3/neurogranin and myelin basic protein messenger ribonucleic acids in the developing rat brain. Endocrinology 141:181–189 [PubMed] [Google Scholar]
- Goldey ES, Kehn LS, Lau C, Rehnberg GL, Crofton KM 1995 Developmental exposure to polychlorinated biphenyls (Aroclor 1254) reduces circulating thyroid hormone concentrations and causes hearing deficits in rats. Toxicol Appl Pharmacol 135:77–88 [PubMed] [Google Scholar]
- Bastomsky CH 1977 Enhanced thyroxine metabolism and high uptake goiters in rats after a single dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Endocrinology 101:292–296 [PubMed] [Google Scholar]
- Crofton KM 2004 Developmental disruption of thyroid hormone: correlations with hearing dysfunction in rats. Risk Anal 24:1665–1671 [PubMed] [Google Scholar]
- Brouwer A, Longnecker MP, Birnbaum LS, Cogliano J, Kostyniak P, Moore J, Schantz S, Winneke G 1999 Characterization of potential endocrine-related health effects at low-dose levels of exposure to PCBs. Environ Health Perspect 107(Suppl 4):639–649 [PMC free article] [PubMed] [Google Scholar]
- Schell LM, Gallo MV, Denham M, Ravenscroft J, DeCaprio AP, Carpenter DO 2008 Relationship of thyroid hormone levels to levels of polychlorinated biphenyls, lead, p,p’- DDE, and other toxicants in Akwesasne Mohawk youth. Environ Health Perspect 116:806–813 [PMC free article] [PubMed] [Google Scholar]
- Herbstman JB, Sjödin A, Apelberg BJ, Witter FR, Halden RU, Patterson DG, Panny SR, Needham LL, Goldman LR 2008 Birth delivery mode modifies the associations between prenatal polychlorinated biphenyl (PCB) and polybrominated diphenyl ether (PBDE) and neonatal thyroid hormone levels. Environ Health Perspect 116:1376–1382 [PMC free article] [PubMed] [Google Scholar]
- Herbstman J, Apelberg BJ, Witter FR, Panny S, Goldman LR 2008 Maternal, infant, and delivery factors associated with neonatal thyroid hormone status. Thyroid 18:67–76 [PubMed] [Google Scholar]
- Goldey ES, Crofton KM 1998 Thyroxine replacement attenuates hypothyroxinemia, hearing loss, and motor deficits following developmental exposure to Aroclor 1254 in rats. Toxicol Sci 45:94–105 [PubMed] [Google Scholar]
- Koibuchi N, Chin WW 2000 Thyroid hormone action and brain development. Trends Endocrinol Metab 11: 123–128 [PubMed] [Google Scholar]
- Nguon K, Baxter MG, Sajdel-Sulkowska EM 2005 Perinatal exposure to polychlorinated biphenyls differentially affects cerebellar development and motor functions in male and female rat neonates. Cerebellum 4:112–122 [PubMed] [Google Scholar]
- Granholm AC 1985 Effects of thyroid hormone deficiency on glial constituents in developing cerebellum of the rat. Exp Brain Res 59:451–456 [PubMed] [Google Scholar]
- Stewart P, Fitzgerald S, Reihman J, Gump B, Lonky E, Darvill T, Pagano J, Hauser P 2003 Prenatal PCB exposure, the corpus callosum, and response inhibition. Environ Health Perspect 111:1670–1677 [PMC free article] [PubMed] [Google Scholar]
- Berbel P, Guadaño-Ferraz A, Angulo A, Ramón Cerezo J 1994 Role of thyroid hormones in the maturation of interhemispheric connections in rats. Behav Brain Res 64:9–14 [PubMed] [Google Scholar]
- Gauger KJ, Kato Y, Haraguchi K, Lehmler HJ, Robertson LW, Bansal R, Zoeller RT 2004 Polychlorinated biphenyls (PCBs) exert thyroid hormone-like effects in the fetal rat brain but do not bind to thyroid hormone receptors. Environ Health Perspect 112:516–523 [PMC free article] [PubMed] [Google Scholar]
- Bansal R, You SH, Herzig CT, Zoeller RT 2005 Maternal thyroid hormone increases HES expression in the fetal rat brain: an effect mimicked by exposure to a mixture of polychlorinated biphenyls (PCBs). Brain Res Dev Brain Res 156:13–22 [PubMed] [Google Scholar]
- Kitamura S, Jinno N, Suzuki T, Sugihara K, Ohta S, Kuroki H, Fujimoto N 2005 Thyroid hormone-like and estrogenic activity of hydroxylated PCBs in cell culture. Toxicology 208:377–387 [PubMed] [Google Scholar]
- Fritsche E, Cline JE, Nguyen NH, Scanlan TS, Abel J 2005 Polychlorinated biphenyls disturb differentiation of normal human neural progenitor cells: clue for involvement of thyroid hormone receptors. Environ Health Perspect 113:871–876 [PMC free article] [PubMed] [Google Scholar]
- Arulmozhiraja S, Morita M 2004 Structure-activity relationships for the toxicity of polychlorinated dibenzofurans: approach through density functional theory-based descriptors. Chem Res Toxicol 17:348–356 [PubMed] [Google Scholar]
- Kimura-Kuroda J, Nagata I, Kuroda Y 2005 Hydroxylated metabolites of polychlorinated biphenyls inhibit thyroid-hormone-dependent extension of cerebellar Purkinje cell dendrites. Brain Res Dev Brain Res 154:259–263 [PubMed] [Google Scholar]
- Bogazzi F, Raggi F, Ultimieri F, Russo D, Campomori A, McKinney JD, Pinchera A, Bartalena L, Martino E 2003 Effects of a mixture of polychlorinated biphenyls (Aroclor 1254) on the transcriptional activity of thyroid hormone receptor. J Endocrinol Invest 26:972–978 [PubMed] [Google Scholar]
- Iwasaki T, Miyazaki W, Takeshita A, Kuroda Y, Koibuchi N 2002 Polychlorinated biphenyls suppress thyroid hormone-induced transactivation. Biochem Biophys Res Commun 299:384–388 [PubMed] [Google Scholar]
- Miyazaki W, Iwasaki T, Takeshita A, Kuroda Y, Koibuchi N 2004 Polychlorinated biphenyls suppress thyroid hormone receptor-mediated transcription through a novel mechanism. J Biol Chem 279:18195–18202 [PubMed] [Google Scholar]
- Frumess RD, Larsen PR 1975 Correlation of serum triodothyronine (T3) and thyroxine (T4) with biologic effects of thyroid hormone replacement in propylthiouracil-treated rats. Metabolism 24:547–554 [PubMed] [Google Scholar]
- Howe S, Borodinsky L, Lyon R 1998 Potential exposure to bisphenol A from food-contact use of epoxy coated cans. J Coat Tech 70:69–74 [Google Scholar]
- Lewis JB, Rueggeberg FA, Lapp CA, Ergle JW, Schuster GS 1999 Identification and characterization of estrogen-like components in commercial resin-based dental restorative materials. Clin Oral Investig 3:107–113 [PubMed] [Google Scholar]
- Schönfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I 2002 Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect 110:A703–A707 [PMC free article] [PubMed] [Google Scholar]
- 1995 Tetrabromobisphenol A and derivatives. Environmental Health Critera no. 172. Geneva: World Health Organization [Google Scholar]
- 1997 Flame-retardants: a general introduction. Environmental Health Critera no. 192. Geneva: World Health Organization [Google Scholar]
- Thomsen C, Lundanes E, Becher G 2002 Brominated flame retardants in archived serum samples from Norway: a study on temporal trends and the role of age. Environ Sci Technol 36:1414–1418 [PubMed] [Google Scholar]
- Moriyama K, Tagami T, Akamizu T, Usui T, Saijo M, Kanamoto N, Hataya Y, Shimatsu A, Kuzuya H, Nakao K 2002 Thyroid hormone action is disrupted by bisphenol A as an antagonist. J Clin Endocrinol Metab 87:5185–5190 [PubMed] [Google Scholar]
- Staples CA, Dorn PB, Klecka GM, O’Block ST, Harris LR 1998 A review of the environmental fate, effects, and exposures of bisphenol A. Chemosphere 36:2149–2173 [PubMed] [Google Scholar]
- Krishnan AV, Stathis P, Permuth SF, Tokes L, Feldman D 1993 Bisphenol-A: an estrogenic substance is released from polycarbonate flasks during autoclaving. Endocrinology 132:2279–2286 [PubMed] [Google Scholar]
- Gaido KW, Leonard LS, Lovell S, Gould JC, Babaï D, Portier CJ, McDonnell DP 1997 Evaluation of chemicals with endocrine modulating activity in a yeast-based steroid hormone receptor gene transcription assay. Toxicol Appl Pharmacol 143:205–212 [PubMed] [Google Scholar]
- Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C, Watson CS, Zoeller RT, Belcher SM 2007 In vitro molecular mechanisms of bisphenol A action. Reprod Toxicol 24:178–198 [PubMed] [Google Scholar]
- Kitamura S, Jinno N, Ohta S, Kuroki H, Fujimoto N 2002 Thyroid hormonal activity of the flame retardants tetrabromobisphenol A and tetrachlorobisphenol A. Biochem Biophys Res Commun 293:554–559 [PubMed] [Google Scholar]
- Zoeller RT, Bansal R, Parris C 2005 Bisphenol-A, an environmental contaminant that acts as a thyroid hormone receptor antagonist in vitro, increases serum thyroxine, and alters RC3/neurogranin expression in the developing rat brain. Endocrinology 146:607–612 [PubMed] [Google Scholar]
- Cheng SY 2005 Thyroid hormone receptor mutations and disease: beyond thyroid hormone resistance. Trends Endocrinol Metab 16:176–182 [PubMed] [Google Scholar]
- Seiwa C, Nakahara J, Komiyama T, Katsu Y, Iguchi T, Asou H 2004 Bisphenol A exerts thyroid-hormone-like effects on mouse oligodendrocyte precursor cells. Neuroendocrinology 80:21–30 [PubMed] [Google Scholar]
- Vermiglio F, Lo Presti VP, Moleti M, Sidoti M, Tortorella G, Scaffidi G, Castagna MG, Mattina F, Violi MA, Crisà A, Artemisia A, Trimarchi F 2004 Attention deficit and hyperactivity disorders in the offspring of mothers exposed to mild-moderate iodine deficiency: a possible novel iodine deficiency disorder in developed countries. J Clin Endocrinol Metab 89:6054–6060 [PubMed] [Google Scholar]
- Siesser WB, Cheng SY, McDonald MP 2005 Hyperactivity, impaired learning on a vigilance task, and a differential response to methylphenidate in the TRβPV knock-in mouse. Psychopharmacology (Berl) 181:653–663 [PubMed] [Google Scholar]
- Ishido M, Masuo Y, Kunimoto M, Oka S, Morita M 2004 Bisphenol A causes hyperactivity in the rat concomitantly with impairment of tyrosine hydroxylase immunoreactivity. J Neurosci Res 76:423–433 [PubMed] [Google Scholar]
- Dowling AL, Martz GU, Leonard JL, Zoeller RT 2000 Acute changes in maternal thyroid hormone induce rapid and transient changes in gene expression in fetal rat brain. J Neurosci 20:2255–2265 [PMC free article] [PubMed] [Google Scholar]
- Dowling AL, Iannacone EA, Zoeller RT 2001 Maternal hypothyroidism selectively affects the expression of neuroendocrine-specific protein A messenger ribonucleic acid in the proliferative zone of the fetal rat brain cortex. Endocrinology 142:390–399 [PubMed] [Google Scholar]
- Dowling AL, Zoeller RT 2000 Thyroid hormone of maternal origin regulates the expression of RC3/neurogranin mRNA in the fetal rat brain. Brain Res Mol Brain Res 82:126–132 [PubMed] [Google Scholar]
- Wu Y, Liu Y, Levine EM, Rao MS 2003 Hes1 but not Hes5 regulates an astrocyte versus oligodendrocyte fate choice in glial restricted precursors. Dev Dyn 226:675–689 [PubMed] [Google Scholar]
- Schuurmans C, Guillemot F 2002 Molecular mechanisms underlying cell fate specification in the developing telencephalon. Curr Opin Neurobiol 12:26–34 [PubMed] [Google Scholar]
- Talsness CE 2008 Overview of toxicological aspects of polybrominated diphenyl ethers: a flame-retardant additive in several consumer products. Environ Res 108:158–167 [PubMed] [Google Scholar]
- Marsh G, Bergman A, Bladh L, Glillner M, Jackobsson E 1998 Synthesis of p-hydroxybromodiphnyl ethers and binding to the thyroid hormone receptor. Organohalogen Compounds (37):305–308 [Google Scholar]
- Newbold RR, Padilla-Banks E, Snyder RJ, Phillips TM, Jefferson WN 2007 Developmental exposure to endocrine disruptors and the obesity epidemic. Reprod Toxicol 23:290–296 [PMC free article] [PubMed] [Google Scholar]
- Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN 2007 Perinatal exposure to environmental estrogens and the development of obesity. Mol Nutr Food Res 51:912–917 [PubMed] [Google Scholar]
- De Ferranti SD, Osganian SK 2007 Epidemiology of paediatric metabolic syndrome and type 2 diabetes mellitus. Diabetes Vasc Dis Res 4:285–296 [PubMed] [Google Scholar]
- Baillie-Hamilton PF 2002 Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med 8:185–192 [PubMed] [Google Scholar]
- Grün F, Blumberg B 2006 Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 147:S50–S55 [PubMed] [Google Scholar]
- Tabb MM, Blumberg B 2006 New modes of action for endocrine-disrupting chemicals. Mol Endocrinol 20:475–482 [PubMed] [Google Scholar]
- Hales CN, Barker DJ 2001 The thrifty phenotype hypothesis. Br Med Bull 60:5–20 [PubMed] [Google Scholar]
- Buresova M, Zidek V, Musilova A, Simakova M, Fucikova A, Bila V, Kren V, Kazdova L, Di Nicolantonio R, Pravenec M 2006 Genetic relationship between placental and fetal weights and markers of the metabolic syndrome in rat recombinant inbred strains. Physiol Genomics 26:226–231 [PubMed] [Google Scholar]
- de Moura EG, Passos MC 2005 Neonatal programming of body weight regulation and energetic metabolism. Biosci Rep 25:251–269 [PubMed] [Google Scholar]
- Cooke PS, Naaz A 2004 Role of estrogens in adipocyte development and function. Exp Biol Med 229:1127–1135 [PubMed] [Google Scholar]
- Wada K, Sakamoto H, Nishikawa K, Sakuma S, Nakajima A, Fujimoto Y, Kamisaki Y 2007 Life style-related diseases of the digestive system: Endocrine disruptors stimulate lipid accumulation in target cells related to metabolic syndrome. J Pharmacol Sci 105:133–137 [PubMed] [Google Scholar]
- Knouff C, Auwerx J 2004 Peroxisome proliferator-activated receptor-γ calls for activation in moderation: lessons from genetics and pharmacology. Endocr Rev 25:899–918 [PubMed] [Google Scholar]
- Kanayama T, Kobayashi N, Mamiya S, Nakanishi T, Nishikawa J 2005 Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor -γ /retinoid X receptor pathway. Mol Pharmacol 67:766–774 [PubMed] [Google Scholar]
- Grün F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, Gardiner DM, Kanno J, Iguchi T, Blumberg B 2006 Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol 20:2141–2155 [PubMed] [Google Scholar]
- Dang ZC, Audinot V, Papapoulos SE, Boutin JA, Löwik CW 2003 Peroxisome proliferator-activated receptor γ (PPAR-γ) as a molecular target for the soy phytoestrogen genistein. J Biol Chem 278:962–967 [PubMed] [Google Scholar]
- Penza M, Montani C, Romani A, Vignolini P, Pampaloni B, Tanini A, Brandi ML, Alonso-Magdalena P, Nadal A, Ottobrini L, Parolini O, Bignotti E, Calza S, Maggi A, Grigolato PG, Di Lorenzo D 2006 Genistein affects adipose tissue deposition in a dose-dependent and gender-specific manner. Endocrinology 147:5740–5751 [PubMed] [Google Scholar]
- Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A 2006 The estrogenic effect of bisphenol-A disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ Health Perspect 114:106–112 [PMC free article] [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H 2004 Global prevalence of diabetes estimates for the year 2000 and projections for 2030. Diabetes Care 27:1047–1053 [PubMed] [Google Scholar]
- Rosenbloom AL, Joe JR, Young RS, Winter WE 1999 Emerging epidemic of type 2 diabetes in youth. Diabetes Care 22:345–354 [PubMed] [Google Scholar]
- Remillard RB, Bunce NJ 2002 Linking dioxins to diabetes: epidemiology and biologic plausibility. Environ Health Perspect 110:853–858 [PMC free article] [PubMed] [Google Scholar]
- Hugo ER, Brandebourg TD, Woo JG, Loftus J, Alexander JW, Ben-Jonathan N 2008 Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ Health Perspect 116:1642–1647 [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Laribi O, Ropero AB, Fuentes E, Ripoll C, Soria B, Nadal A 2005 Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic α-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environ Health Perspect 113:969–977 [PMC free article] [PubMed] [Google Scholar]
- Poirier P, Després JP 2003 Waist circumference, visceral obesity, and cardiovascular risk. J Cardiopulm Rehabil 23:161–169 [PubMed] [Google Scholar]
- Gardner JD, Brower GL, Voloshenyuk TG, Janicki JS 2008 Cardioprotection in female rats subjected to chronic volume overload: synergistic interaction of estrogen and phytoestrogens. Am J Physiol Heart Circ Physiol 294:H198–H204 [PubMed] [Google Scholar]
- Deodato B, Altavilla D, Squadrito G, Campo GM, Arlotta M, Minutoli L, Saitta A, Cucinotta D, Calapai G, Caputi AP, Miano M, Squadrito F 1999 Cardioprotection by the phytoestrogen genistein in experimental myocardial ischaemia-reperfusion injury. Br J Pharmacol 128:1683–1690 [PMC free article] [PubMed] [Google Scholar]
- Chan YH, Lau KK, Yiu KH, Li SW, Chan HT, Tam S, Shu XO, Lau CP, Tse HF 2007 Isoflavone intake in persons at high risk of cardiovascular events: implications for vascular endothelial function and the carotid atherosclerotic burden. Am J Clin Nutr 86:938–945 [PubMed] [Google Scholar]
- de Kleijn MJ, van der Schouw YT, Wilson PW, Grobbee DE, Jacques PF 2002 Dietary intake of phytoestrogens is associated with a favorable metabolic cardiovascular risk profile in postmenopausal U.S. women: the Framingham study. J Nutr 132:276–282 [PubMed] [Google Scholar]
- Cerami C, Founds H, Nicholl I, Mitsuhashi T, Giordano D, Vanpatten S, Lee A, Al-Abed Y, Vlassara H, Bucala R, Cerami A 1997 Tobacco smoke is a source of toxic reactive glycation products. Proc Natl Acad Sci USA 94:13915–13920 [PMC free article] [PubMed] [Google Scholar]
- Koschinsky T, He CJ, Mitsuhashi T, Bucala R, Liu C, Buenting C, Heitmann K, Vlassara H 1997 Orally absorbed reactive glycation products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci USA 94:6474–6479 [PMC free article] [PubMed] [Google Scholar]
- Sandu O, Song K, Cai W, Zheng F, Uribarri J, Vlassara H 2005 Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes 54:2314–2319 [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Piperi C, Korkolopoulou P, Kandaraki E, Levidou G, Papalois A, Patsouris E, Papavassiliou AG 2007 Accumulation of dietary glycotoxins in the reproductive system of normal female rats. J Mol Med 85:1413–1420 [PubMed] [Google Scholar]
- Uribarri J, Stirban A, Sander D, Cai W, Negrean M, Buenting CE, Koschinsky T, Vlassara H 2007 Single oral challenge by advanced glycation end products acutely impairs endothelial function in diabetic and nondiabetic subjects. Diabetes Care 30:2579–2582 [PubMed] [Google Scholar]
- McLachlan JA 2006 Commentary: prenatal exposure to diethylstilbestrol (DES): a continuing story. Int J Epidemiol 35:868–870 [PubMed] [Google Scholar]
- Lang IA, Galloway TS, Scarlett A, Henley WE, Depledge M, Wallace RB, Melzer D 2008 Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA 300:1303–1310 [PubMed] [Google Scholar]
- Emmen JM, McLuskey A, Adham IM, Engel W, Verhoef-Post M, Themmen AP, Grootegoed JA, Brinkmann AO 2000 Involvement of insulin-like factor 3 (Insl3) in diethylstilbestrol-induced cryptorchidism. Endocrinology 141:846–849 [PubMed] [Google Scholar]
- Ben Rhouma K, Tébourbi O, Krichah R, Sakly M 2001 Reproductive toxicity of DDT in adult male rats. Hum Exp Toxicol 20:393–397 [PubMed] [Google Scholar]
- Fisher JS, Macpherson S, Marchetti N, Sharpe RM 2003 Human testicular dysgenesis syndrome: a possible model using in-utero exposure of the rat to dibutyl phthalate. Hum Reprod 18:1383–1394 [PubMed] [Google Scholar]
- Parks LG, Ostby JS, Lambright CR, Abbott BD, Klinefelter GR, Barlow NJ, Gray Jr LE 2000 The plasticizer diethylhexyl phthalate induces malformations by decreasing fetal testosterone synthesis during sexual differentiation in the male rat. Toxicol Sci 58:339–349 [PubMed] [Google Scholar]
- Gupta C 2000 Reproductive malformation of the male offspring following maternal exposure to estrogenic chemicals. Proc Soc Exp Biol Med 224:61–68 [PubMed] [Google Scholar]
- Timms BG, Howdeshell KL, Barton L, Bradley S, Richter CA, vom Saal FS 2005 Estrogenic chemicals in plastic and oral contraceptives disrupt development of the fetal mouse prostate and urethra. Proc Natl Acad Sci USA 102:7014–7019 [PMC free article] [PubMed] [Google Scholar]
- Ramos JG, Varayoud J, Sonnenschein C, Soto AM, MuñozDe Toro M, Luque EH 2001 Prenatal exposure to low doses of bisphenol A alters the periductal stroma and glandular cell function in the rat ventral prostate. Biol Reprod 65:1271–1277 [PubMed] [Google Scholar]
- Cohn BA, Cirillo PM, Wolff MS, Schwingl PJ, Cohen RD, Sholtz RI, Ferrara A, Christianson RE, van den Berg BJ, Siiteri PK 2003 DDT and DDE exposure in mothers and time to pregnancy in daughters. Lancet 361:2205–2206 [PubMed] [Google Scholar]
- Markey CM, Wadia PR, Rubin BS, Sonnenschein C, Soto AM 2005 Long-term effects of fetal exposure to low doses of the xenoestrogen bisphenol-A in the female mouse genital tract. Biol Reprod 72:1344–1351 [PubMed] [Google Scholar]
- Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenbergh JG, vom Saal FS 1999 Exposure to bisphenol A advances puberty. Nature 401:763–764 [PubMed] [Google Scholar]
- Honma S, Suzuki A, Buchanan DL, Katsu Y, Watanabe H, Iguchi T 2002 Low dose effect of in utero exposure to bisphenol A and diethylstilbestrol on female mouse reproduction. Reprod Toxicol 16:117–122 [PubMed] [Google Scholar]
- Zsarnovszky A, Le HH, Wang HS, Belcher SM 2005 Ontogeny of rapid estrogen-mediated extracellular signal-regulated kinase signaling in the rat cerebellar cortex: potent nongenomic agonist and endocrine disrupting activity of the xenoestrogen bisphenol A. Endocrinology 146:5388–5396 [PubMed] [Google Scholar]
- Steinberg RM, Walker DM, Juenger TE, Woller MJ, Gore AC 2008 The effects of perinatal PCBs on adult female rat reproduction: development, reproductive physiology, and second generational effects. Biol Reprod 78:1091–1101 [PMC free article] [PubMed] [Google Scholar]
- Jenkins S, Rowell C, Wang J, Lamartiniere CA 2007 Prenatal TCDD exposure predisposes for mammary cancer in rats. Reprod Toxicol 23:391–396 [PMC free article] [PubMed] [Google Scholar]
- Mizuyachi K, Son DS, Rozman KK, Terranova PF 2002 Alteration in ovarian gene expression in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin: reduction of cyclooxygenase-2 in the blockage of ovulation. Reprod Toxicol 16:299–307 [PubMed] [Google Scholar]
- Newbold R 1995 Cellular and molecular effects of developmental exposure to diethylstilbestrol: implications for other environmental estrogens. Environ Health Perspect 103(Suppl 7):83–87 [PMC free article] [PubMed] [Google Scholar]
- Kaufman RH 1982 Structural changes of the genital tract associated with in utero exposure to diethylstilbestrol. Obstet Gynecol Annu 11:187–202 [PubMed] [Google Scholar]
- Krstevska-Konstantinova M, Charlier C, Craen M, Du Caju M, Heinrichs C, de Beaufort C, Plomteux G, Bourguignon JP 2001 Sexual precocity after immigration from developing countries to Belgium: evidence of previous exposure to organochlorine pesticides. Hum Reprod 16:1020–1026 [PubMed] [Google Scholar]
- Blanck HM, Marcus M, Tolbert PE, Rubin C, Henderson AK, Hertzberg VS, Zhang RH, Cameron L 2000 Age at menarche and Tanner stage in girls exposed in utero and postnatally to polybrominated biphenyl. Epidemiology 11:641–647 [PubMed] [Google Scholar]
- Martin OV, Lester JN, Voulvoulis N, Boobis AR 2007 Forum: human health and endocrine disruption: a simple multicriteria framework for the qualitative assessment of end point-specific risks in a context of scientific uncertainty. Toxicol Sci 98:332–347 [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Piperi C, Patsouris E, Korkolopoulou P, Panidis D, Pawelczyk L, Papavassiliou AG, Duleba AJ 2007 Immunohistochemical localization of advanced glycation end-products (AGEs) and their receptor (RAGE) in polycystic and normal ovaries. Histochem Cell Biol 127:581–589 [PubMed] [Google Scholar]