Endocrinology
E ndocrinology is the scientific and medical discipline that focuses on hormones, including the function and disorders of endocrine glands.
The endocrine system consists of several glands that secrete hormones that affect the different organ systems (Figure1).
The main endocrine glands are the hypothalamus, pituitary gland, thyroid gland, parathyroid gland, adrenal glands, pancreas, and gonads (ovary and testis).
Figure 1. Main endocrine glands and their functions
Disorders of the Hypothalamus and Pituitary Gland
O f the endocrine glands, the hypothalamus and pituitary glands are of major importance since they act as the coordinating centers of the endocrine system.
The hypothalamus is responsible for maintaining the body’s internal balance (homeostasis) by stimulating or inhibiting major bodily functions such as the heart rate and blood pressure, body temperature, fluid and electrolyte balance, appetite and body weight, sleep cycle and function of the gastrointestinal track. The hypothalamus is also considered the master regulator of the endocrine system; Regulatory hormones secreted by the hypothalamus are transported by the hypophyseal-portal system to the anterior and posterior pituitary (Figure 2), prompting the release of secondary hormones that can affect various organ functions.
Hormones typically bind to membrane or nuclear receptors of specific target glands/organs, which in turn release hormones that exert negative feedback control on the releasing sites. (Figure 3).
Figure 3. Example of negative (in red) feedback inhibition:
The hypothalamus secretes releasing factors that act on the pituitary gland to stimulate the release of trophic hormones.
Trophic hormones act then on target organs (e.g., adrenal, thyroid or gonads), which in response produce other
hormones/signals, shutting down the production of releasing or/and trophic hormones.
The hypothalamus secretes various hormones that are then transported to the anterior pituitary (Figure 4):
- Corticotropin releasing hormone (CRH)
- Growth hormone releasing hormone (GHRH)
- Thyrotropin releasing hormone (TRH)
- Gonadotropin releasing hormone (GnRH)
- Somatostatin (inhibits growth hormone secretion)
- Prolactin releasing and prolactin inhibitor hormones
Figure 4. Overview of hypothalamic and pituitary hormones and their actions
The pituitary gland also secretes hormones in response to hypothalamic release hormones:
- Thyroid stimulating hormone (TSH)
- Adrenocortical stimulating hormone (ACTH)
- Follicle stimulating hormone (FSH)
- Luteinizing hormone (LH)
- Prolactin
- Melanocyte-stimulating hormone
- Endorphins
Supraoptic and paraventricular nuclei of the hypothalamus also secrete hormones that are transported to the posterior pituitary to be released into the circulation. These are:
Disorders of the hypothalamus can result in appetite, temperature and sleep disorders. As an example, hypothalamic obesity occasionally develops in response to major hypothalamic injury/damage affecting the centers of appetite regulation and energy balance. Hypothalamic obesity is characterized by an uninhibited eating disorder that often results in morbid obesity and can be associated with other obesity complications like diabetes, dyslipidemia, obstructive sleep apnea, mood disorder etc.
Disorders of the hypothalamus and/or anterior pituitary can also result in hypopituitarism, including adrenal insufficiency (see adrenal disorders section), hypothyroidism (see thyroid disorders section), hypogonadism (see puberty and its disorders section), growth hormone deficiency (see growth disorders section) and prolactin deficiency (inability to lactate).
Disorders of the posterior pituitary can result in diabetes insipidus and disorders pertaining to oxytocin deficiency (like inability to lactate, vaginal dryness, decreased libido etc.).
Diabetes insipidus
T he neurohypophysis, or posterior pituitary gland, secretes vasopressin (AVP), also known as anti-diuretic hormone (ADH). AVP is synthesized by the supraoptic and paraventricular nuclei of the hypothalamus (see picture below), in response to plasma osmolality, intravascular blood volume changes (like bleeding, third spacing, etc.) and non-osmotic stimuli (e.g., nausea, drugs).
Figure 5. Physiology of the Hypothalamus and Posterior Pituitary
The antidiuretic effect of ADH is regulated through V2, cAMP dependent- receptors and aquaporing-2 proteins inducing increased water permeability and increased urea movement on the collecting ducts. In addition, ADH increases the rate of absorption of sodium (NaCl) in the thick ascending loop of Henle.
Etiology: Central DI or nephrogenic DI can be complete or partial, familial or acquired (e.g. brain tumor in central DI and lithium in nephrogenic DI).
Symptoms: polyuria and polydipsia, enuresis, FTT/poor growth, unexplained fevers, constipation
Diagnosis: elevated serum Na in patients with abnormal thirst mechanism and dilute urine (as above) vs failure to concentrate urine after a water deprivation test in patients with normal Na/ intact thirst mechanism
Treatment: monitor/maintain fluid balance (drink water as needed) and use ADH-agonists to prevent nocturia and minimize sleep disruption. ADH agonist include L-arginine Vasopressin (natural AVP-subcutaneous), DDAVP (synthetic- intranasal, IV or subcutaneous, or oral) and thiazide diuretics.
Syndrome of inappropriate secretion of ADH (SIADH)
SIADH is due to excessive ADH secretion producing inappropriate urinary concentration and water retention, resulting in euvolemic hyponatremia.
Etiology: CNS disorders (head injury, CNS tumors, hydrocephalus etc.), pulmonary conditions (infections, cystic fibrosis), ectopic production (lung cancer etc.) drugs (thiazides etc.).
Symptoms: headaches, nausea, seizures, focal deficits
Diagnosis: low serum Na, low serum Osm, absence of signs of hypovolemia like tachycardia or hypervolemia like edema and ascites, absence of hypoadrenalism or hypothyroidism (that can also cause euvolemic hyponatremia)
Treatment: fluid restriction and drugs (e.g. vaptans- block ADH binding to V2 receptors)
Growth disorders
P hysical growth refers to bodily changes that happen as a child matures including weight, length or height and head circumference increases. Growth failure is by far one of the most common reasons for referrals to the pediatric endocrine office.
Linear growth is most rapid in prenatal life when it is mainly regulated by maternal and placental factors. Postnatal growth is progressively slower and predominantly reflective of childs own genetic potential (see growth velocity figure). Another growth acceleration occurs at puberty.
Measurements
Accurate measurement can not be overemphasized in the evaluation of the short child.
Height and Length:
Growth Velocity: Growth velocity (cm/yr) = (Height2-Height1) / (#months between times) x 12. It determines normal or abnormal growth by comparing height change over a period of time to gender-appropriate norms.
Body Proportions:
- Arm Span (AS) is the distance from one end of child’s arm (measured at the fingertips) to the other. AS is measured fingertip to fingertip while standing flat against a wall with arms outstretched. At birth, AS is less than length by about 2.5 cm. By 10 years of age, AS is equal to height; and after 10 years in boys and 12 years in girls, AS exceeds height by up to 5 cm. If AS is greater than 5 cm, pathologic causes of tall stature such as Marfan’s Syndrome or hypogonadism should be considered.
- Upper to lower segment ratio (U/L) reflects trunk vs legs ratio, where lower segment is the distance from the middle of pubic symphysis to the floor level and upper segment is height minus lower segment. At birth, U/L is about 1.7:1 or trunk longer than legs. U/L then decreases by 0.1 for every year of age until 10 years of age when it becomes 1:1 (trunk=legs). After 10 years, the ratio is
- Weight to length ratio (w/l) or BMI (Body Mass Index) is useful to assess pediatric overweight and obesity. BMI is calculated by dividing the patient’s weight in kilograms by their height in meters squared. A BMI between the 85th to 95th percentiles is defined as overweight, and a BMI greater than the 95th percentile defines obesity. It is also informative for the differential diagnosis of short stature (e.g., w/l >1 and with short stature may suggest endocrinopathy, w/l
Figure 6. Measuring Upper to Lower segment ratio
Mid-parental height (MPH): measure of child’s genetic height potential, using parents adult heights.
Mid parental height calculation:
- Males = [mother’s height (in) + father’s height (in) + 5 inches]/2
- Females = [mother’s height (in) + father’s height (in) – 5 inches]/2
- Normal genetic height range is MPH +/- 3.5
Bone Age x-ray: tool for assessment of skeletal maturation as it compares to the chronological age and for the prediction of child’s final adult height (for films older than age 6 years). Bone age x-ray is typically delayed in thyroid hormone and growth hormone deficiency or constitutional delay of growth, normal in familial short stature and advanced in precocious puberty.
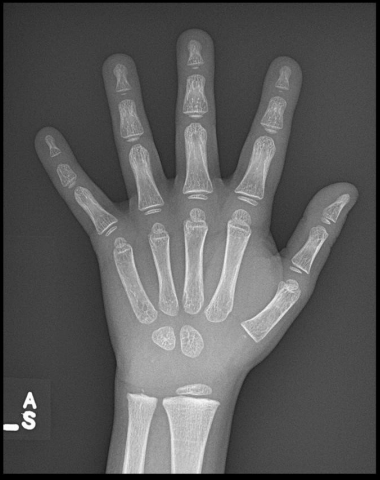
Figure 7. Example of bone age x-ray.
Children < the 3rd %ile on the growth chart or >2 standard deviations below MPH deserve a short stature evaluation.
Short stature etiology
C ommon parental concerns about height tend to be gender biased (e.g. boys that are the shortest in their class or girls that are “too tall” or “taller than even her male classmates”). Referral for a short stature evaluation is also commonly prompted by concerns over bullying/ teasing at school for child’s size/stature.
Normal variants
- Familial short stature. Parents are short; therefore, their children are likely to be short. Growth velocity is typically normal along with body proportions, labs and bone age. However, since some pathological causes of short stature are familial, an evaluation may be warranted for children who are very short on the growth chart despite growing along their genetic target height trajectory
- Constitutional growth delay (“late blooming”). Children’s growth velocity is usually normal in childhood but progressively declines and it can be associated with delayed puberty. However, catch-up growth occurs without assistance and children eventually achieve heights within their genetic target height range (although typically below their mid-parental height). Bone age x-ray is typically delayed for chronological age.
Pathological causes
- Nutritional deficiency: Undernutrition (global nutritional deprivation or marasmus, inadequate protein intake or Kwashiorkor, anorexia nervosa, zinc deficiency)
- Psychosocial dwarfism: Extreme emotional deprivation can result in failure to thrive (both poor height and weight growth) or even short stature with normal BMI.
- IUGR or small for gestational age: Poor growth due to maternal, placental or chromosomal factors. About 10% of children born SGA fail to “catch-up” to normal growth percentiles by age 2 years.
- Systemic disease: malabsorption, heart disease, renal disease, heme/onc disease, pulmonary disease, diabetes mellitus, inborn errors of metabolism, chronic infection, inflammatory disease
- Chromosomal abnormalities/genetic syndromes: Turner Syndrome, Prader-Willi Syndrome, Russel Sliver Syndrome, Noonan Syndrome
- Skeletal dysplasias: achondroplasia or hypochonodroplasia etc.
- Endocrinopathy: thyroid hormone deficiency, growth hormone deficiency, Cushing Syndrome
- Chronic drug intake: supra physiologic glucocorticoid exposure, high-dose estrogens or androgens, stimulent medications (e.g., methylphenidate, dextroamphetamine)
- Idiopathic short stature: Height < 2 SD below the corresponding mean height for a given age, sex and population group without evidence of systemic, endocrine, nutritional or chromosomal abnormalities
Diagnosis: Bone age x-ray (to assess skeletal maturation), CBC, CMP, ESR, UA to screen for systemic disease, free T4, TSH, IGF-1, and IGFBP-3 to screen for endocrinopathy. Celiac screen optional, Pituitary/brain MRI as needed. Karyotype/microarray or specialized genetic testing as clinically indicated.
Typically, growth hormone replacement is reserved for true growth hormone deficiency. Many causes of short stature are due to an underlying disease and by treating this disease, you are simultaneously able to treat the short stature. For example, levothyroxine treatment for hypothyroidism and growth hormone for growth hormone deficiency typically restores linear growth. However, growth hormone treatment is also FDA approved for Turner Syndrome, small-for-gestational age with failure to catch up, Prader-Willi Syndrome, idiopathic short stature, SHOX gene haploinsufficiency, Noonan Syndrome and chronic kidney disease.
Tall stature is defined as predicted adult stature greater than two standard deviations above the mean height for age and gender. Tall stature can represent a normal variant of growth such as familial tall stature or it may be pathological.
- growth hormone excess
- Disorders of pubertal development
- familial glucocorticoid deficiency
- Hyperthyroidism
- Hyperinsulinism
- Pituitary (i.e. MEN type 1, McCune-Albright
- Ectopic growth hormone
- Growth hormone releasing hormones tumors
- precocious puberty
- Obesity
- T1DM
- IDM
- Persistent hyperinsulinemic hypoglycemia of infancy
- Lipodystrophy
- Beckwith-Wiedemann
- Sotots Syndrome
- Weaver Syndrome
- Beckwith-Widemann
- Perlman Syndrome
- Simpson-Golabi-Behemel Type1
- Proteus
- Nevo
- Marfan
- Homcystinuria
- Fragile X
Table 2. Common causes of pahological tall stature
Diagnosis: Bone age x-ray, thyroid function tests, fasting insulin level
Thyroid disorders
T hyroid hormone is very important for the regulation of body metabolism and thermogenesis throughout life but it is of most critical importance for CNS development in the fetal and postnatal periods (especially the first 3 years of life). Thyroid hormone production is under the control of hypothalamic TRH and pituitary TSH but also iodine availability.
Figure 8. Phathophysiological Aspects of Thyroid Hormone Disorders/thyroid Peroxidase Autoantibodies and Reproduction
Fetal thyroid development: The fetal thyroid gland arises from an outpouching of the foregut and develops at the base of the tongue from where it will migrate down to its permanent location (over the thyroid cartilage) between the 4-8th weeks of gestation. It begins to concentrate iodine at about 10 weeks and hypothalamopituitary axis control is established at 18 weeks. Fetal T4 and TSH gradually rise to reach adult levels by 36 weeks of gestation. The rise in fetal serum T3 is less pronounced due to placental and fetal deiodeinases that covert T4 to the inactive hormone reverse T3 (rT3).
In response to the stress of birth and ambient temperature changes in the first 24 hours of life, TSH peaks up to 80 mIU/mL, driving T4 and T3 increases. Then all thyroid indices gradually normalize within the next several weeks (typically 4-6). Prematurity and perinatal complications may affect this normal process, resulting in delayed TSH rise or lower than expected T3/T4 levels.
Hypothyroidism (low thyroid hormone secretion)
Etiology: congenital vs acquired.
Congenital hypothyroidism (CH) is most commonly secondary to dysplasia/ hypoplasia of the thyroid gland or ectopia. In a minority of newborns, hypothyroidism is secondary to a defect in the synthesis of thyroid hormones (dyshormonogenesis), CNS/hypothalamic disorders affecting TRH or TSH synthesis, exposure to maternal antithyroid medication or as a result from transplacental transfer of TSH-receptor blocking antibodies. Outside the United States, the most common etiology of CH is iodine deficiency (or endemic goiter). Iodization of salt has significantly reduced CH in the United States.
Acquired hypothyroidism is most commonly due to Hashimoto thyroiditis, a destructive autoimmune disease in which there is lymphocytic infiltration of the thyroid gland. Other causes include post-surgical hypothyroidism (due to thyroid gland removal for hyperthyroidism or thyroid nodules/cancer), sick euthyroid syndrome, and subacute / suppurative thyroiditis. In autoimmune hypothyroidism (as also seen in autoimmune hyperthyroidism), children typically have a history or family history of autoimmune disease.
Most newborns with CH appear normal at birth, unless mother was hypothyroid in which case babies may present with signs and symptoms of CH including jaundice (due to indirect hyperbilirubinemia), dry and coarse skin, umbilical hernia, constipation, thick protuberant tongue, poor muscle tone, anemia, hoarseness, growth retardation, delayed ossification and delayed closure of the anterior fontanel. Thyroid gland enlargement (goiter) is not usually present unless there is dyshormonogenesis. If babies with CH remain untreated, intellectual disability may ensue.
Children and teens with acquired hypothyroidism may present with decreased linear growth or even stunted growth, cold intolerance, constipation, dry skin, hair thinning or hair loss, sleepiness or irregular periods. A goiter and slow deep tendon reflexes may be present on exam. Delayed bone age maturation is also typically seen.
- Newborn screen in the United States (TSH, T4 or combination of TSH and T4 depending on the state program)
- Thyroid function studies typically show an elevated TSH and low free and total T4 or T3.
- Thyroid ultrasound may demonstrate a hypoplastic or absent thyroid gland.
- Nuclear medicine thyroid scan may also be indicated in cases of suspected ectopic gland or dyshormonogenesis. In cases of autoimmune hypothyroidism thyroid peroxidase and thyroglobulin antibodies may be positive.
Treatment includes levothyroxine. Dose and monitoring schedule is variable based on age of child. For newborns, dose is ~ 10-15 mcg/kg/day. Goal of treatment is to keep free T4 levels above the mean reference range with suppressed TSH.
Hyperthyroidism (excessive thyroid hormone secretion)
Etiology. The most common etiology is Graves’ disease (autoimmune stimulatory condition). Other rare causes include neonatal thyrotoxicosis due to maternal Graves’ disease, activating TSH receptor mutation, and thyrotoxicosis as seen in the context of McCune-Albright Syndrome. Hot thyroid nodules are very rare in pediatrics.
Symptoms: include weight loss despite hyperphagia, palpitations, heat intolerance, increased perspiration, fatigue, diarrhea, hand tremor, anxiety and restlessness, goiter, rapid linear growth, irregular menses, and exophthalmos.
Treatment: anti-thyroid drugs, radiation ablation (RAI) therapy or thyroidectomy. Beta- blockade is also typically necessary to decrease cardiac work.
Thyroid nodules
Thyroid nodules are uncommon (incidence 1-1.5 %). Risk factors include female sex, pubertal age, medial or family history of thyroid disease. These typically present with an asymptomatic movable, soft and non-tender thyroid mass.
Thyroid cancer
Thyroid cancer rare in children, typically presents with a painless or tender thyroid mass that is hard and fixated and associated with painless palpable neck lymphadenopathy. Distant metastases (neck, lungs, bones) are frequently present at the time of diagnosis, but survival/prognosis is excellent if early diagnosis and appropriate treatment are established. The most common type is papillary thyroid carcinoma (PTC). Follicular carcinoma is the second most common type of and is more prevalent in iodine-deficient regions. Medullary thyroid carcinomas are very rare and also the most aggressive, occurring most commonly as part of the familial MTC syndromes (e.g. MEN type 2A, MEN 2B, Carney complex etc.). The biggest risk factor is radiation exposure outside the familial cases.
Adrenal disorders (cortex and medulla)
T he adrenal gland is divided into twodistinct areas, the cortex and the medulla. The cortex is further divided in 3 zones (zona glomerulosa or site of aldosterone synthesis, zona fasciculata or site of cortisol synthesis and zona reticularis or site of androgen synthesis). The medulla serves as the site of catecholamine, metanephrine and normetanephrine synthesis.
Figure 9. Adrenal glandareas and zones with respective hormones synthesized
Cholesterol (low-density lipoprotein or LDL) is the precursor for adrenal steroidogenesis. Adrenal steroidogenesis includes three main pathways, leading to cortisol, aldosterone and androgen synthesis, with androgens eventually being aromatized (converted to estrogens). See figure 10 below, where arrows represent enzymes that convert one substrate to another.
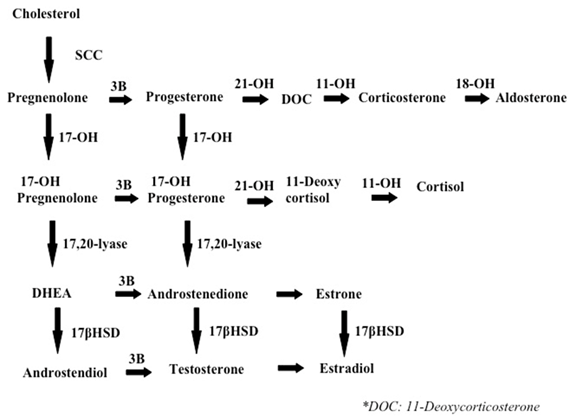
Figure 10: Main pathways of adrenal steriodogenesis
- Cortisol secretion is under the control of the hypothalamic CRH and pituitary ACTH. Cortisol production itself also shuts down the system at the pituitary and hypothalamic levels (negative feedback inhibition). Cortisol actions affect almost every body system (Figure 11 below).
- Aldosterone secretion is under the control of the renin-angiotensin system (not under the control of the brain). Aldosterone regulates water retention, Na and K balance and body’s blood pressure.
- Androgens are important for sexual development and reproduction in both males and females. Androgens also serve as precursors of estrogens.
- Catecholamines are neurotransmitters that regulate various central nervous system functions, but are also hormones that regulate cardiovascular and respiratory function, secretions and muscle contraction.
Figure 11: Multiple systemic effects of cortisol
Cortisol excess (or Cushing Syndrome)
Etiology: most commonly exogenous (iatrogenic or due to supraphysiologic exposure to glucocorticoids); rarely endogenous. Endogenous is most commonly due to excess pituitary ACTH secretion (Cushing disease or ACTH producing pituitary adenoma) and rarely due to ectopic ACTH production (e.g. lung or thymic tumor) or excess cortisol secretion by an adrenal tumor.
Symptoms: growth stunting, central obesity, facial plethora, buffalo hump, sleep disorders, hypertension, bruising, tinea corporis, and wide striae
Diagnosis: confirm hypercortisolism with low dose dexamethasone test vs midnight salivary cortisol vs 24-hr urinary free cortisol x3 days, if high cortisol check serum ACTH and refer to endocrinology for more specialized testing and imaging
Treatment: surgical removal of tumor, supplementary radiotherapy for Cushing disease
Adrenal insufficiency
Etiology: most commonly iatrogenic (treatment with suprpaphysiologic doses of glucocorticoids can result to adrenal suppression) vs primary (adrenal gland is malfunctioning) vs central (pituitary or and hypothalamus are malfunctioning). Primary adrenal insufficiency can be autoimmune, due to congenital adrenal hyperplasia, adrenoleukodystrophy, adrenal hemorrhage etc. Central is due to isolated ACTH deficiency or associated with multiple pituitary deficiencies.
Symptoms: hypoglycemia, nausea, emesis, fatigue, anorexia, hypotension, hyponatremia, salt craving and hyperkalemia (if aldosterone deficiency), decreased pubic and axillary hair (if adrenal androgen deficiency), hyperpigmentation (due to high MSH).
Diagnosis: hyponatremia and hyperkalemia, high PRA, low aldosterone, elevated ACTH, failed ACTH stimulation test
Treatment: acute phase (IVF, IV hydrocortisone), chronic phase (glucocorticoids and fludrocortisones)
Congenital adrenal hyperplasia
(discussed in detail under the ambiguous genitalia section)
Pheochromocytoma
Etiology: Catecholamine-secreting tumors, arising from chromaffin cells of the adrenal medulla. Very rare in children and usually inherited (MEN 2b, MEN 2a, NF-1, VHL, familial pheo/para etc).
Symptoms: flushing, sweating, palpitations, hypertension, headaches, emesis, and feeling of impending death
Diagnosis: Plasma or urine fractionated catecholamines/meta- and normetanephrines, CT adrenals and PET-FDG scan
Treatment: Removal of tumor with pre-op preparation to include alpha and beta blockade
Ambiguous genitalia and disorders of sexual development (DSD)
S ex determination and sex differentiation can be divided into a) chromosomal sex or karyotype (46 XX, 46 XY, or variants), b) gonadal sex (presence of a testis or ovary) and c) phenotypic sex (external genitalia and internal structures).
Human embryo is bipotential with genetic sex determined at conception. Male differentiation requires specific molecular and biochemical stimuli at specific times. Female differentiation occurs in absence of male determinant genes.
Disorders of sexual development can be associated with ambiguous genitalia (i.e., external genitals neither appearing clearly male nor female).
Disorders in androgen synthesis or action
Other (i.e. hypospadias, congenital HH, cryptorchidism)
- Klinefelter Syndrome
- Turner Syndrome
- Mixed gonadal dysgenesis
- Congenital adrenal hyperplasia (CAH)
- Androgen insensitivity syndrome
- Cryptorchidism
Klinefelter Syndrome (1 in 500-1000 live births)
Etiology: 47, XXY (classic) vs 46, XY/47, XXY (mosaic)
Symptoms: Tall stature, gynecomastia, small testes, azoospermia, learning difficulties
Diagnosis: High FSH and LH, low AMH and inhibin B, testosterone low in 50-75% of cases
Treatment: Testosterone replacement, spontaneous fertility unlikely
Turner Syndrome (1 in 2500 live births) and mixed gonadal dysgenesis
Etiology: 45 X (classic) vs Mosaic types (or mixed gonadal dysgenesis) like 45, XO/46, XX; 45, XO/46, XY etc.
Symptoms: Lymphedema, webbed neck/low hairline, high-arched palate, shield chest/wide-spaced nipples, short stature, streak ovaries, coarctation of aorta or other CV defects, learning difficulties
Diagnosis: prenatal typically. Based on cardiac anomalies, short stature in childhood, delayed puberty (high FSH & LH) in puberty.
Treatment: Estrogen/progesterone replacement therapy, long-term follow-up (cardiovascular, bone, reproductive health, hearing), spontaneous fertility unlikely, gonadectomy only when Y fragment contains TSPY locus (risk of gonadoblastoma increased)
Congenital Adrenal Hyperplasia (CAH)
Etiology: Group of autosomal recessive disorders, most common type is due to CYP21A2 gene mutations resulting in 21-hydroxylase deficiency. Classic (1:15,000 births; impaired cortisol synthesis). Non-classic (1:100- 1:1,000; cortisol synthesis relatively normal). Less common types include 11-beta hydroxylase deficiency, 17-hydroxylase deficiency, 3-beta- hydroxysteroid dehydrogenase deficiency.
Symptoms: in 21-hydroxylase deficiency, cortisol and aldosterone are not produced therefore patient present with adrenal salt/salt wasting symptoms. Also due to excess ACTH, excessive androgens are produced that can cause virilization of female infants or milder symptoms of hyperandrogenemia (like acne, premature pubarche, irregular periods) in the non-classic form. In 11-beta hydroxylase and 17-hydroxylase deficiency there is HTN due to excess mineralocorticoid and in 3-beta-hds there is adrenal insufficiency and androgen insufficiency, therefore males are under masculinized.
Figure 13 – Female infant with ambiguous genitalia (clitoromegaly, bifid scrotum, rugated folds) due to classic 21-hydroxylase deficiency.
Diagnosis: In 21-hydroxylase deficiency, newborn screen (NBS) detects most cases. If no NBS in place, classic type can present with hyponatremia, hyperkalemia, low aldosterone, high PRA, high 17-hydroxy progesterone, high androstenedione. With the non-classic type there is typically high serum 17-hydroxy progesterone, androstenedione and testosterone.
Management: For classic 21-hydroxylase deficiency: glucocorticoids, mineralocorticoids and also salt for infants. For non-classic 21-hydroxylase deficiency, glucorticoids may be needed but not always (depends on degree of hyperandrogenemia symptoms).
Androgen insensitivity syndrome
Most common cause of male undermasculinization
Etiology: 46, XY/ X-linked recessive/ Androgen Receptor (AR) gene mutation
Symptoms: Wide phenotypic spectrum:
- Complete androgen receptor resistance (CAIS): female phenotype with BL inguinal hernias or with 1o amenorrhea & scanty pubic hair
- Partial androgen receptor resistance (PAIS): ambiguous genitalia with labial masses or male with infertility, decreased pubic hair or beard growth and gynecomastia
Diagnosis: age-appropriate testosterone levels, elevated LH & FSH, normally formed testes, absent/vestigial Mullerian structures, variable presence of Wolffian ducts
Treatment: hormone replacement therapy +/- gonadectomy
Cryptorchidism
Cryptorchidism mean that the testis is not in the scrotum and is not descended by 4 months old. This is the most common congenital abnormality in boys, present in 2-4% of term male infants. Complications include inguinal hernia, testicular torsion or trauma, subfertility, malignant transformation. Management involves orchidopexy (surgically moving the testes into the scrotum) or removal of dysgenic testis. Hormonal therapy (hCG/GnRH) may help testicular descent.
Puberty
N ormal female puberty. The hypothalamus-pituitary-ovarian (HPO) axis gradually increases in activity from the prepubertal age causing increases in LH, FSH, estradiol, inhibit A, and Inhibin B. These changes will eventually result in ovarian estrogen production and as a result breast growth, nipple and areola development, female body habitus, cornification of the vaginal epithelium, and enlargement of the uterus.
Normal male puberty. The increasing rise of LH and FSH causes an increase in testosterone, estrone, estradiol, androstenedione, 17-hydroxyprogesterone, Inhibin A, and DHEA. LH and FSH act directly on the testes to stimulated Leydig and Sertoli cell proliferation. Testicular testosterone causes enlargement of the penis, broadening of the shoulders with narrowing of the hips, deepening of the voice, beard growth, temporal hair recession, and thick/curly truncal hair.
Assessment: Tanner staging (or sexual maturation staging) by inspection and palpation is used to assess the sexual maturity of the child.
Figure 14. Tanner Stages
from https://openi.nlm.nih.gov/detailedresult.php?img=PMC4478390_fnhum-09-00344-g001&req=4
The onset of puberty in girls is defined by thelarche (breast budding) and it is typically between 8 and 12 years of age, although it can be as early as age 6 years in African American females. Puberty onset in boys is defined by gonadarche (testicular enlargement) and it is typically between 9 to 14 years of age. This is based upon data of predominately Caucasian children, but recent statistics reveal that other ethnicities may have earlier onsets of puberty. *
* Reference: AAP and Pediatric Endocrine Society, Precocious Puberty: A Guide for Families. 2012. https://www.aap.org/en-us/Documents/soen_precocious_puberty.pdf
Adrenarche: increased adrenal androgen production measured by DHEAS. It precedes gonadarche and pubarche.
Gonadarche: increased activity of the HPO axis
Pubarche: onset of sexual hair growth
Thelarche: onset of breast development
Menarche: onset of first menstrual period
Disorders of Puberty
Precocious puberty
Precocious puberty is when the physical changes of puberty (thelarche with other secondary sex characteristics for girls, and gonadarche for boys) begin before the lower age limit for that sex, which is < 8 years for girls and < 9 years for boys. The lower limit is lower for other ethnicities such as African American or Mexican-American. Causes of precocious puberty can be gonadotropin dependent or gonadotropin independent.
Incomplete precocious puberty
GnRH-Dependent or Central
Secondary to chronic exposure to sex steroids
Space occupying or pressure associated lesions
CAH, Gonadotropin-independent puberty, McCune-Albright
Adrenal sex steroid secreting, Gonadotropin-producing, Ovarian, Testicular
Chronic primary hypothyroidism, CAH, Exogenous sex steroid or gonadotropins, Ovarian cysts
Table 4: Etiologies of Precocious Puberty
Diagnosis: Evaluation includes suggestive physical exam findings, imaging in the form of bone age x-ray (typically advanced for chronological age), MRI of the head, pelvic/testicular ultrasound, and labs including LH, FSH, estradiol, testosterone, DHEAS, Free T4, TSH, and a GnRH stimulation test.
Treatment: includes treating the underlying cause, and if the cause is gonadotropin-dependent, then GnRH agonist therapy may be appropriate.
Delayed puberty
Delayed puberty is defined as lack of testicular enlargement by age 14 in boys and lack of thelarche by 13 in girls, no menarche by age 16, or no pubertal progression for 3 years after the onset of puberty. Delayed puberty can be due to central etiology (hypogonadotropic hypogonadism) or peripheral etiology (hypergonadotropic hypogonadism). Delayed puberty is most commonly due to “functional” hypogonadotropic hypogonadism such as in constitutional delay of growth and puberty, chronic illness, anorexia nervosa, etc. Pathological causes of delayed puberty can be divided into categories of primary hypothalamic-pituitary dysfunction and primary gonadal disorder.
Etiologies of Pubertal Delay
- Congenital (i.e. GnRH deficiency without or without anorexia, part of anterior pituitary hormone deficiencies, midline craniofacial anomalies)
- Acquired (i.e. tumors, trauma, marijuana, infiltration, CDP, chronic systemic disease, malnutrition, diabetes mellitus, hypothyroidism, hyperprolactinemia, Cushing’s disease, anorexia nervosa)
Diagnosis: Evaluation includes a physical examination, bone age XR, FSH/LH/testosterone/estradiol levels, cranial MRI, pelvic ultrasound, and karyotyping or genetic studies.
Treatment: short course of IM testosterone to trigger (“jump start”) puberty in boys, oral estrogen/progesterone for puberty induction in girls. Long-term hormone replacement therapy may be needed in both sexes based on the nature of the problem and possibly continued androgen replacement therapy. Treatment for girls includes cyclic estrogen and progesterone.
Mineral disorders
C alcium, phosphorus, vitamin D, and the hormones that regulate them are very important in skeletal development and mineralization. Abnormalities in these can disrupt calcium homeostasis and therefore cause disorders of mineral metabolism or skeletal disorders of childhood.
Calcium homeostasis
Figure 15. Low serum calcium stimulates release of PTH from the parathyroid glands. PTH stimulates calcium release
from bone and the kidneys to reabsorb calcium. The kidneys also excrete phosphorus and produce more vitamin D.
Vitamin D stimulates intestinal absorption of calcium.
Hypocalcemia
- Exaggeration of physiologic decrease in serum calcium
- Insufficient PTH due to immature parathyroid hormone or immature kidney response; more commonly seen in premature infants
- Late onset more frequently associated with hyperphosphatemia
- Early – birth to 3 days of life
- Late – onset at 1 week of life
- Congenital hypoparathyroidism
- Maternal hypercalcemia
- Phosphorus overload from cow’s milk formula
- Uremia
- Congenital Vitamin D deficiency
- Improper parathyroid gland formation
- Destruction of parathyroid glands
- Impaired parathyroid gland function
- Parathyroid and thymus hypoplasia: DiGeorge Syndrome
- Parathyroid dysgenesis: Kenny-Caffey Syndrome and Sanjad-Sakati Syndrome
- Parathyroid aplasia or dysplasia: inactivation of GCM2 gene
- APS type 1: triad of hypoparathyroidism, adrenal insufficiency, and mucocutaneous candidiasis
- PTH gene mutations
- Anti-CaSR antibodies
- Mitochondrial disease
Diagnosis: Measure serum calcium and vitamin-25OHD with corresponding phosphorus and PTH. Also, magnesium and alkaline phosphatase should also be checked. An ECG should be obtained to evaluate for changes related to hypocalcemia (i.e. prolonged QT interval).
- Acute management: IV bolus infusions of calcium gluconate with continuous infusion of elemental calcium
- Chronic management: Oral calcium, calcitriol, and a low phosphorus formula (Similac PM 60/40).
Hypercalcemia
Hypercalcemia with elevated PTH, normal phosphorus, normal magnesium, elevated alkaline phosphatase, and low urinary calcium
Facial anomalies, cardiac and renal defects, hyperacusis, and cognitive impairment
Elevated calcium, elevated PTH, low phosphorus, and elevated calcitriol
Elevated serum calcium and urinary calcium
Osteoclastic activity leading to excessive calcium release from bone
Diagnosis: Measure calcium, phosphorus, PTH, magnesium, Vitamin-25OHD, and alkaline phosphatase. ECG will show shortened ST segment and heart block.
Treatment: Increase urinary excretion of sodium with furosemide. Start calcitonin to indirectly inhibit osteoclastic bone resoprtion or inhibit bone resorption directly with bisphosphonates.
Metabolic Bone Disease
Rickets occurs when there is defective mineralization of the epiphyseal growth plate. Although there are different causes of rickets, all forms show characteristic widening and flaring of the epiphyses.
- Exclusively breast-fed infants for at least 6 months
- Lack of exposure to direct sunlight
- GI disorders: biliary atresia, CF, IBD, and celiac disease
- Liver disease
- Drugs that induce CYP450 enzymes
- X-linked hypophosphatemia
- Autosomal dominant/recessive hypophosphatemic rickets
- Hereditary hypophosphatemic rickets with hypercalciuria
Table 8. Etiologies of Metabolic Bone Disease.
Disorders of the endocrine part of pancreas
G lucose is an important source for brain energy metabolism and extensive regulatory mechanisms are in place to ensure protection from hypoglycemia. Glucose concentrations naturally reach a nadir a couple hours after birth and then begin to rise reaching normal values by day 3 of life. This is related to the abrupt cessation of placental glucose transfer at delivery causing a transient decrease in glucose levels with subsequent response of increased glucagon, decreased insulin levels, and an increase in catecholamine secretion to gradually normalize plasma glucose concentration. Regulatory mechanisms in older children are balanced by gluconeogenesis and glycogenolysis. Hypoglycemia definition varies based on the age group (40 mg/dL or below in neonates) and < 55-60 mg/dL in older children.
Glucose Homeostasis
Figure 16. This figure describes the balance of glucose regulatory mechanisms.
Lower blood glucose levels stimulate the pancreas to release glucagon to stimulate glycogen breakdown in the liver to increase blood glucose levels.
High blood glucose levels stimulate insulin release from the pancreas to stimulate glycogen formation in the liver to lower blood glucose levels.Hypoglycemia
Etiology: Hypoglycemia is due to defects of the hormones or enzymes of the glucose regulatory mechanisms that result in inadequate glucose or surplus of insulin.
- Hyperinsulinism (i.e. infant of diabetic mother, erythroblastosis fetalis, Beckwith-Wiedeman, Nesideroblastosis)
- Metabolic (i.e. Maple syrup urine disease, G6PD, debrancher enzyme deficiency, liver phosphorylase deficiency, glycogen synthetase deficiency, defects in fatty acid oxidation)
- Hormonal (i.e. panhypopituitarism, adrenal insufficiency, growth hormone deficiency)
Table 9. Etiologies of Hypoglycemia by Age
Symptoms: An abrupt decrease in plasma glucose causes adrenergic symptoms (due to catecholamine release) such as pallor, sweating, tachycardia, tremor, and emesis. A slow decrease in plasma glucose causes neuroglycopenic symptoms such as confusion, diplopia, headache, dizziness, lethargy, seizure, and lack of coordination. Neonatal symptoms of hypoglycemia are more subtle, such as apnea, low temperature, poor tone, jitteriness, and poor feeding.
Treatment: Acute treatment includes early feeding (neonates), dextrose gel, or IV boluses of 2 mL/kg of 10% dextrose in water followed by continuous infusions of glucose. If hypoglycemia persists, it is then treated with continuous glucose infusion, diazoxide and/or somatostatin analogs. If hypoglycemia persists, pancreatectomy may be necessary.
Diabetes Mellitus
Diabetes mellitus (DM) is a common metabolic condition of hyperglycemia caused by complete or partial insulin deficiency and its actions.
Insulin physiology: Insulin is made on the ribosomes of pancreatic islet beta cells as a proinsulin precursor (single chain: chain A is connected to chain B by c-peptide) that is then cleaved into insulin and c-peptide molecule. It is then released into circulation as a double chain polypeptide linked by disulfide bridges. C-peptide has a longer half-life and later nadir.
Etiologies: There are many subgroups with distinct etiologies but the most commonly known will be discussed.
Type 1 DM (previously referred to as Juvenile Onset Diabetes): T1DM is due to autoimmune destruction of pancreatic beta cells resulting in insulin deficiency. Histocompatibility locus antigens that determine response to self or other antigens create antibodies to cytoplasmic and cell surface components of the islets cells, to insulin, to GAD (glutamic acid decarboxylase or enzyme in pancreatic islet innervation), and to IA-2 (islet phosphatase). T1DM is the most common type in childhood and adolescence (7-15 year olds); but with the epidemic increases in childhood obesity, there is an increased prevalence in T2DM among this age. Wasting and diabetic ketoacidosis are complications from undiagnosed/ untreated hyperglycemia.
Type 2 DM: T2DM is typically due to insulin resistance with some B-cell impairment. The insulin resistance can be observed at the level of skeletal muscle, liver, and adipose tissue, but can result in actual insulin deficiency. For more info regarding the differences between T1DM and T2DM see table below:
Healthy nutrition, excercise, oral agents
Table 10. Comparison of Type 1 and Type 2 Diabetes Mellitus.
Monogenic diabetes (MODY or maturity onset of diabetes of youth). MODY has a dominant inheritance pattern with the mutations of the genes encoding enzymes or transcription factors affecting islet cell development and insulin secretion. This causes GLUT2 transporter regulation to be mutated.
Symptoms: polyuria, polydipsia, weight loss (T1DM); obesity (T2DM) and acanthosis nigricans. Complications: retinopathy, nephropathy, neuropathy, ischemic heart disease, vasculopathy, diabetic ketoacidosis, nonketotic hyperosmolar coma.
Diagnosis: Initial laboratory workup includes random BG or fasting BG, HbA1C, UA. BMP and blood gas are typically obtained if there is concern for DKA. Celiac panel, thyroid function tests, c-peptide and autoimmune diabetes antibodies studies (i.e. anti-GAD, anti-islet, etc) are obtained if T1DM is suspected.
Laboratory findings between T1DM and T2DM
Table 11. Laboratory findings for T1DM and T2DM
Kussmaul respirations, oriented but sleepy
Kussmual or depressed respirations; sleep to depressed sensorium or coma
Table 12. Classification of DKA (from Nelson Textbook of Pediatrics)
T1DM: Patients are typically dependent on exogenous insulin; fluids; diet.
T2DM: First line treatment entails a diabetic diet (healthy, sugar avoidance, and CHO restriction) coupled with increased physical exercise. Oral hypoglycemic are typically introduced at the time of diagnosis. Exogenous insulin treatment is reserved for severe forms that have had DKA or HgA1C > 9%.
Obesity: Disorders of Energy Balance
O besity is defined as BMI > 95 th percentile for age and sex. Overweight is defined as BMI of 85-95 th percentile. Increasingly common in children (US prevalence 18.5% or ~ 13.7 million children and adolescents). Obesity is associated with risk for comorbidities that can persist into adulthood, such as diabetes mellitus, hypertension, early onset cardiovascular disease and osteoarthritis, dyslipidemia, precocious puberty, polycystic ovarian syndrome etc.
Etiology: most commonly exogenous (usually due to a combination of genetic predisposition and dietary/lifestyle habits). Other etiologies include drug-induced and endocrine-related. Genetic conditions associated with obesity include Prader-Willi Syndrome, Alstrom Syndrome, Bardet-Bied Syndrome, Down Syndrome, and Turner Syndrome. Endocrinopathies that can be associated with obesity include Cushing Syndrome and hypothalamic obesity and less likely growth hormone deficiency or hypothyroidism. Antidepressants, antiepileptics, glucocorticoids, and atypical antipsychotics are associated with an increased risk of obesity.
Diagnosis: Based on physical exam/ BMI. Screening for co-morbid conditions as sleep apnea, non-alcoholic fatty liver disease, type 2 diabetes, hypertension, dyslipidemia, or polycystic ovary syndrome is imperative.
Treatment: dietary/lifestyle changes to promote weight loss. The only FDA approved medication for obesity in children less than 16 years of age is Orlistat but has unfavorable side effects, such as flatulence and oily, loose stools. Bariatric surgery can be considered for children with BMI greater than 40 kg/m 2 with near-complete skeletal maturity.
Pediatric Dyslipidemia
L ipids are organic and water insoluble compounds and include fatty acids, triglycerides, and cholesterol. Lipoproteins (LDL, IDL, VLDL, HDL, and chylomicrons) are soluble lipid or fat proteins that facilitate lipid movement within the aqueous environment of the body.
After digested fat is absorbed, chylomicrons are produced in the intestinal lumen and then transferred to adipose tissue, muscle, and the liver. There chylomicrons will be hydrolyzed to free fatty acids or VLDL that in turn will be metabolized to LDL (the major carrier of cholesterol to tissues). HDL acts as a “bad fat” scavenger, transferring excess cholesterol from peripheral tissues to cells that require it or to the liver to be excreted. Any abnormalities in these pathways or metabolism can lead to lipid disorders.
Etiology: The prevalence of dyslipidemia in children age 12-19 was reported as approximately 20.3% in the CDC Morbidity and Mortality Weekly Report. The most common etiology of dyslipidemia is exogenous (usually due to excessive simple carbohydrate and saturated fat intake with limited physical activity). Other etiologies include genetic (familial hypertriglyceridemia, familial hypercholesterolemia, familial defective apoB100, familial combined hyperlipidemia etc.), drug-induced (glucocorticoids, isotretinoin, protease inhibitors, immunosuppressives, beta blockers), and endocrinopathies (hypothyroidism, hyperthyroidism, T2DM, Cushing Syndrome).
Symptoms: usually associated with obesity and acanthosis nigricans. Hypertriglyceridemia may be associated with lipemia retinalis and cutaneous eruptive xanthomata. Hypercholesterolemia can be associated with xanthelasmas, corneal arcus, and tuberous xanthomata.
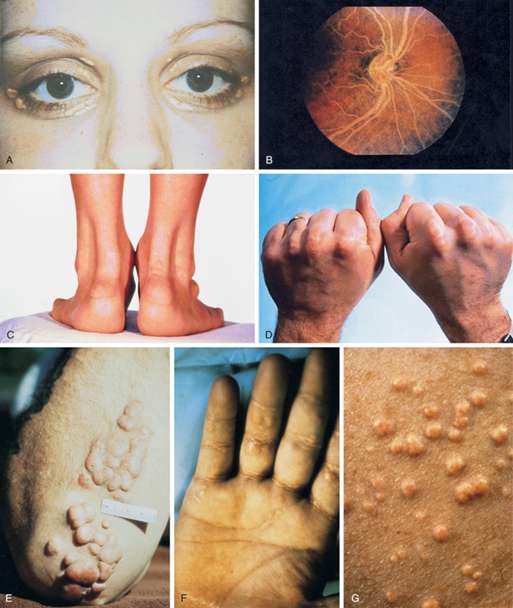
Figure 18 – Physical examination findings associated with hyperlipidemia.
From Williams Textbook of Endocrinology by Shlomo Melmed, pg. 1678.
A, Xanthelasma. B, Lipemia retinalis. C, Achilles tendon xanthomas. Notice the marked thickening of the tendons.
D, Tendon xanthomas. E, Tuberous xanthomas. F, Palmar xanthomas. G, Eruptive xanthomas.Diagnosis: Universally screen children aged 9-11 years with a non-fasting or fasting lipid profile. A diagnosis of dyslipidemia is made with total cholesterol greater than 169 mg/dL, LDL greater than 109 mg/dL, non-HDL greater than 119 mg/dL, Apo-B greater than 89 mg/dL, triglycerides greater than 74 mg/dL in children aged 0-9 years or greater than 89 mg/dL in children older than 9 years old, or HDL less than 45 mg/dL
Treatment: diet (CHILD-1 and CHILD-2) and lifestyle changes is the mainstay. Statins are considered for cases of severe hypercholesterolemia or moderate hypercholesterolemia with risk factors/comorbidities if there is no response to a minimum of 6 months of diet and lifestyle intervention.
Polyendocrinopathies
T here are syndromes with a constellation of multiple endocrine diseases termed polyendocrinopathies.