Biochemistry of adipose tissue: an endocrine organ
1 Ciências Químicas e das Biomoléculas e Unidade de Mecanismos Moleculares da Doença do Centro de Investigação em Saúde e Ambiente, Escola Superior de Tecnologia da Saúde do Porto, Instituto Politécnico do Porto, Portugal
2 Centro de Farmacologia e Biopatologia Química (U38-FCT), Faculdade de Medicina da Universidade do Porto, Portugal
Teresa Oliveira
2 Centro de Farmacologia e Biopatologia Química (U38-FCT), Faculdade de Medicina da Universidade do Porto, Portugal
Ruben Fernandes
1 Ciências Químicas e das Biomoléculas e Unidade de Mecanismos Moleculares da Doença do Centro de Investigação em Saúde e Ambiente, Escola Superior de Tecnologia da Saúde do Porto, Instituto Politécnico do Porto, Portugal
2 Centro de Farmacologia e Biopatologia Química (U38-FCT), Faculdade de Medicina da Universidade do Porto, Portugal
1 Ciências Químicas e das Biomoléculas e Unidade de Mecanismos Moleculares da Doença do Centro de Investigação em Saúde e Ambiente, Escola Superior de Tecnologia da Saúde do Porto, Instituto Politécnico do Porto, Portugal
2 Centro de Farmacologia e Biopatologia Química (U38-FCT), Faculdade de Medicina da Universidade do Porto, Portugal
Corresponding author: Prof. Ruben Fernandes, ESTSP, Rua Valente Perfeito 322, 4400-330 Vila Nova de Gaia, Portugal. Phone: + 351 222 061 004. E-mail: tp.ppi.pstse@fpr
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Adipose tissue is no longer considered to be an inert tissue that stores fat. This tissue is capable of expanding to accommodate increased lipids through hypertrophy of existing adipocytes and by initiating differentiation of pre-adipocytes. Adipose tissue metabolism exerts an impact on whole-body metabolism. As an endocrine organ, adipose tissue is responsible for the synthesis and secretion of several hormones. These are active in a range of processes, such as control of nutritional intake (leptin, angiotensin), control of sensitivity to insulin and inflammatory process mediators (tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), resistin, visfatin, adiponectin, among others) and pathways (plasminogen activator inhibitor 1 (PAI-1) and acylation stimulating protein (ASP) for example). This paper reviews some of the biochemical and metabolic aspects of adipose tissue and its relationship to inflammatory disease and insulin resistance.
Adipose tissue
Originally considered as simply a storage organ for triacylglycerol, interest in the biology of adipose tissue has increased substantially. Over the last decades there has been considerable accumulation of experimental data about the biology and biochemistry of adipose tissue. This tissue is no longer considered to be an inert tissue that just stores fat [1]. Adipose tissue is a metabolically dynamic organ that is the primary site of storage for excess energy but it serves as an endocrine organ capable of synthesizing a number of biologically active compounds that regulate metabolic homeostasis. This dynamic tissue is composed not only of adipocytes, but also of other cell types called the stroma-vascular fraction, comprising blood cells, endothelial cells, pericytes and adipose precursor cells among others [2–5]. Several studies have evidenced that adipose tissue is not uniform. Depending on the location in the body, they differ in their capacity to secrete adipocytokines, as well as cellular composition with varied phenotype, as well as the quantity and proportion of adipocytes forming it, blood vessel stromal cells and immune system cells [6]. It is now generally recognized that adipose tissue is an important organ of a complex network that participates in the regulation of a variety of quite diverse biological functions ( Figure 1 ) [7–10].
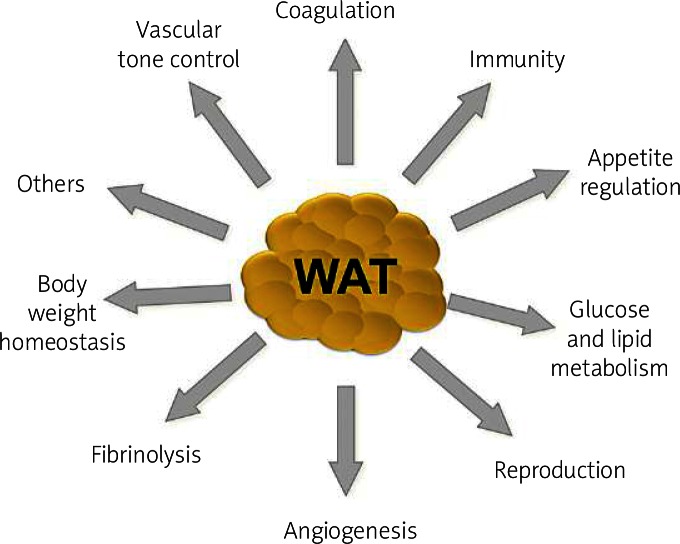
The most significant physiological functions of white adipose tissue such as coagulation, appetite regulation, immunity, glucose and lipid metabolism, reproduction, angiogenesis, fibrinolysis, body weight homeostasis and vascular tone control
Adipogenesis refers to the differentiation of pre-adipocytes into mature fat cells, i.e. the development of adipose tissue, which varies according to sex and age. Adipocytes differentiate from stellate or fusiform precursor cells of mesenchymal origin. The morphological and functional changes that take place in the course of adipogenesis correspond to a shift in transcription factor expression and activity leading from a primitive, multipotent state to a final phenotype characterized by alterations in cell shape and lipid accumulation [4, 5].
Pre-adipocytes within adipose tissue can differentiate into mature adipocytes throughout life, thus enabling hyperplastic expansion of adipose tissue when increased storage requirements are needed. In addition, the mature adipocytes can expand in size to accommodate increased storage needs and in situations of overnutrition become hypertrophic. As a result, adipocyte number and morphology transform in response to energy balance via the biochemical processes involved in lipid uptake, esterification, lipolysis and differentiation of pre-adipocytes [11].
In mammals, there are two types of adipose tissue: white and brown. The adipocytes in these two types exhibit different morphology and function.
Brown adipose tissue specialized in heat production (thermogenesis) is almost absent in adult humans, but is found at birth. Brown adipocytes, with an average diameter, are smaller than adipocytes of white adipose tissue. They have a number of cytoplasmic lipid droplets of different sizes, cytoplasm relatively abundant, a spherical core and slightly eccentric and numerous mitochondria that release heat by oxidation of fatty acids. Brown adipose tissue also stores energy in lipid form, but more regularly produces heat by oxidizing fatty acids within the adipocyte, rather than supplying free fatty acids for use by other cell types [2, 4, 5]. Brown fat derives its color from extensive vascularization and the presence of many densely packed mitochondria. It is traversed by many more blood vessels than white fat. These blood vessels assist in delivering fuel for storage and oxidation, and in dispersing heat generated by the numerous mitochondria to other parts of the body [8, 9].
Although its participation in thermogenesis is irrelevant, white adipose tissue’s functional capacity is much broader and more comprehensive. It has extensive distribution in the body, involving, or infiltrating, almost the entire region subcutaneously by organs and hollow viscera of the abdominal cavity or mediastinum and several muscle groups, for which it offers mechanical protection, softening the impact of shocks and allowing appropriate sliding of muscle bundles, one on the other, without compromising their functional integrity [2, 4]. Because it is an excellent thermal insulator and has a wide distribution, including the dermis and subcutaneous tissue, it plays an important role maintaining body temperature [5]. By this ability to accumulate and provide energy when necessary, it assumes the status of the most important buffering system for lipid energy balance, particularly fatty acids, which are an exceptionally efficient fuel storage species. The highly reduced hydrocarbon tail can be readily oxidized to produce large quantities of ATP (adenosine triphosphate) [9].
Lipogenesis and lipolysis
Fat accumulation is determined by the balance between fat synthesis (lipogenesis) and fat breakdown (lipolysis/fatty acid oxidation).
Lipogenesis is a process that occurs preferentially in adipose tissue ( Figure 2 ), but it also happens in liver, and it is the synthesis of fatty acids, which are used as energy reserves. This process is responsive to changes in the diet [12]. It is stimulated by a high carbohydrate diet leading to elevated postprandial plasma triglyceride levels, whereas lipogenesis is inhibited by polyunsaturated fatty acids and by fasting. Fasting is related to a decrease in plasma glucose and an increase in plasma-free fatty acids. These effects are partly mediated by hormones, which inhibit (leptin) or stimulate (angiotensin, acylation stimulating protein) lipogenesis. Glucose itself is a substrate for lipogenesis. It increases the process by stimulating the release of insulin and inhibiting the release of glucagon from the pancreas [12].
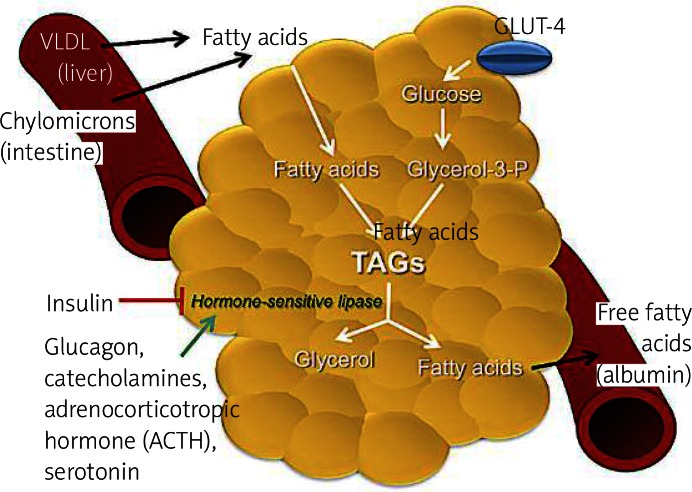
Primary metabolic role of adipose tissue. In the feeding state, insulin-dependent glucose transport 4 (GLUT 4) allows the uptake of glucose from the bloodstream to adipocytes. Glycolysis occurs, producing glycerol-3-phosphate (glycerol-3-P), a substrate required for lipogenesis. Fatty acids from liver carried by very low-density lipoproteins (VLDL) and chylomicrons from the intestine are esterified with glycerol-3-P to form lipid droplets of triacylglycerols (TAGs). In the fasting state and in stress conditions, hormonesensitive lipase is activated for lipolysis. Some steps are required to produce glycerol, which travels to the liver, and fatty acids. These free fatty acids will tra vel in the bloodstream to the liver, muscle and to other organs to be oxidized. In the bloodstream fatty acids are immediately bound to albumin
Lipolysis occurs in adipose tissue and is the breakdown of fat, in other words, from energy reserves (triglycerides) for energy production by which triacylglycerol molecules are hydrolyzed to free fatty acids and glycerol ( Figure 2 ). During times of metabolic stress (i.e. during fasting or prolonged arduous exercise when the body’s energy needs exceed the circulating nutrient levels), the adipocyte’s triacylglycerol droplet is degraded to provide free fatty acids to be used as an energy source by other tissues. Numerous stimuli are able to elicit the lipolytic response in adipocytes. However, ultimately the same pair of enzymes, hormone-sensitive lipase and monoacylglycerol lipase, is responsible for catalyzing the hydrolysis of the triacylglycerol ester bonds. Complete hydrolysis of triacylglycerol involves the breakage of 3 ester bonds to release free fatty acids and a glycerol moiety. The same enzyme, hormone-sensitive lipase, is responsible for facilitating hydrolysis of the esters at positions 1 and 3 of the triacylglycerol. A second enzyme, 2-monoacylglycerol lipase, catalyzes hydrolysis of the remaining ester to yield a third free fatty acid and glycerol. Hormone-sensitive lipase is inhibited by insulin and is favored by the presence of glucagon and epinephrine [9, 10, 12]. Glycerol is effluxed out of adipocytes via an aquaporin type of transport molecule and must be shuttled back to the liver for use in oxidation or gluconeogenesis. However, under maximal lipolytic conditions, substantial recycling of fatty acids occurs such that on average about two fatty acid molecules are released per glycerol molecule. Outside the adipocyte, fatty acids are immediately bound to albumin and carried in the bloodstream to the liver, muscle and other tissues for oxidation [10].
β-Oxidation is a catabolic process in which the free fatty acids resulting from lipolysis are used by the body as a source of energy. The fatty acid molecules are converted into acetyl coenzyme A molecules [2].
Secretory organ
Through the discovery of the ability to secrete hormones, great importance has been attributed to the role of adipose tissue [3].
White adipose tissue may represent the largest endocrine tissue of humans. Its pleiotropic nature is based on the ability of fat cells to secrete numerous hormones, growth factors, enzymes, cytokines, complement factors and matrix proteins. Adipose tissue also expresses receptors for most of these factors that are implicated in the regulation of many processes including food intake, energy expenditure, metabolism homeostasis, immunity and blood pressure homeostasis [7, 13].
Adipose tissue is dynamically involved in cell function regulation through a complex network of endocrine (signals travel through the circulatory system to reach all parts of the body), paracrine (signals sent only to cells in the vicinity of the cell station), and autocrine (only affecting cells that are the same type) signals that influence the response of many tissues, including hypothalamus, pancreas, liver, skeletal muscle, kidneys, endothelium, and the immune system, among others. This secretory nature has prompted the view of white adipose tissue as an extremely active endocrine tissue [5].
It is commonly assumed that under normal physiological circumstances adult humans are practically devoid of functional brown adipose tissue [14]. However, it has been recently shown that human white adipose tissue can be infiltrated with brown adipocytes expressing uncoupling protein 1 (UCP-1). This protein is found in the mitochondria of brown adipose tissue and it generates heat by non-shivering thermogenesis.
Experimental data suggest that there are some differences, in respect to adipokine synthesis and secretion, between visceral fat and subcutaneous adipose tissue, as visceral fat appears to be more active. Both types of this tissue are characterized by production of a unique profile of adipocytokines. In the visceral tissue, for example, higher concentrations of IL-6 (interleukin-6) and PAI-1 (plasminogen activator inhibitor 1) are observed. In turn, in the subcutaneous tissue, there is a higher concentration of leptin and adiponectin [6].
The endocrine activity of white adipose tissue was postulated when its capacity for steroid hormone interconversion was alluded to. Particularly since the discovery of leptin, in 1994, the list of adipocyte-derived factors has been increasing at an extraordinary pace. The discovery of leptin opened up a whole field of studies into the biology of adipocytes, their metabolic and endocrine functions, and the functional relationships between secretions of adipocytes and peripheral metabolic functions. A different way of addressing the production of adipose-derived factors is by focusing on the function they perform [4, 5].
The adipose tissue secretes a number of bioactive substances ( Figure 3 ), such as adipocytokines among others. Unbalanced production of pro- and anti-inflammatory adipocytokines in obese adipose tissue may contribute to many aspects of the metabolic syndrome (MetS).
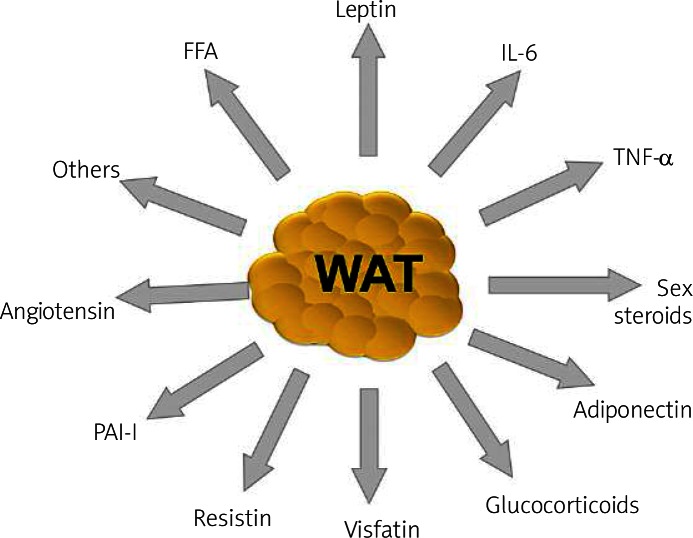
Some of the factors secreted by white adipose tissue, which underlie the multifunctional nature of this endocrine organ: adiponectin, leptin, angio tensin, resistin, visfatin, acylation stimulating protein (ASP), sex steroids, glucocorticoids, tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), and free fatty acids (FFA), among others
Oversecretion of potentially harmful adipocytokines, such as PAI-1, tumor necrosis factor-α (TNF-α) or visfatin, and hyposecretion of potentially beneficial adipocytokines, such as adiponectin, might be major mechanisms involved in lifestyle-related diseases, including diabetes mellitus, hyperlipidemia, hypertension and atherosclerosis, comprising the so-called MetS [15, 16]. The reduction of visceral fat therefore might be an essential preventive measure for MetS and its consequence, cardiovascular disease. The regulation of key adipocytokines such as adiponectin might be considered as an efficient therapeutic procedure, but needs to be studied carefully [13].
Obesity is now viewed as a state of systemic, chronic low-grade inflammation [17]. It has been recognized by recent studies that obesity (waist circumference) has a strong impact on adipokine secretion and insulin resistance [18]. More recently it has been recognized that macrophages are an important part of the secretory function of adipose tissue and the main source of inflammatory cytokines, such as TNF-α and IL-6. As a secretory organ, adipose tissue presents several particularities and its secretory activity is regulated by humoral and hormonal mechanisms ( Table I ).
Table I
Factors secreted by adipose tissue into the bloodstream and respective function/effect in their targets
| Molecule | Function/effect |
|---|---|
| Leptin | Signals to the brain about body fat stores. Regulation of appetite and energy expenditure. Wide variety of physiological functions |
| Adiponectin | Plays a protective role in the pathogenesis of type 2 diabetes and cardiovascular disease |
| Resistin | Hypothetical role in insulin resistance |
| TNF-α | Affects insulin receptor signaling, possible cause of the development of insulin resistance in obesity |
| IL-6 | Pro-inflammatory, lipid and glucose metabolism, regulation of body weight |
| PAI-1 | Inhibitor of the fibrinolytic system by inhibition of activation of plasminogen |
| Angiotensinogen | Precursor of angiotensin II; regulator of blood pressure and electrolyte homeostasis |
| FFA | Oxidized in tissues to produce local energy. Serve as a substrate for triglyceride and structural molecular synthesis. Involved in the development of insulin resistance |
| ASP | Influences the rate of triacylglycerol synthesis in adipose tissue |
| VEGF | Stimulation of angiogenesis |
| Adipsin | Potential relation between the complement pathway and adipose tissue metabolism |
| Glycerol | Structural component of the major classes of biological lipids and gluconeogenic precursor |
| IGF-1 | Stimulates proliferation of a wide variety of cells and mediates many cells and many of the effects of growth hormone |
TNF-α – tumor necrosis factor α, IL-6 – interleukin-6, PAI-1 – plasminogen activator inhibitor 1, FFA – free fatty acids, ASP – acylation stimulating protein, VEGF – vascular endothelial growth factor, IGF-1 – insulin-like growth factor 1
Leptin
Leptin is a small peptide (16 kDa), considered as a pre-inflammatory cytokine that indicates common structural and functional properties, belonging to the IL-6 family of cytokines [17, 19, 20]. The ob gene expressed by adipocytes encodes it. It is an anorexigenic peptide that increases energy expenditure, and is primarily cleared from plasma by the kidney through glomerular filtration followed by proteolytic degradation in the renal tubules. The leptin receptor is expressed not only in the central nervous system, but also in some peripheral tissues (hematopoietic and immune cells), suggesting that leptin may have functions other than food intake and energy expenditure regulation [17, 19]. Adipose tissue and plasma leptin concentrations are dependent on the amount of energy stored as fat as well as the status of energy balance. Therefore, leptin levels are higher in obese individuals and increase with overfeeding. Conversely, lean individuals have lower leptin levels and fasting results in reduction of circulating leptin. Nutritional regulation of leptin is mediated at least in part by insulin, as leptin decreases in response to low insulin levels and increases with feeding or in response to insulin stimulation [10].
Leptin is expressed mainly by adipose tissue, although low levels have been detected in the placenta, skeletal muscle, gastric and mammary epithelium and the brain. Leptin is increased by glucocorticoids, acute infection and proinflammatory cytokines. In contrast, cold exposure, adrenergic stimulation, growth hormone (GH), thyroid hormone, melatonin, smoking and thiazolidinediones decrease leptin [12, 13]. Its levels are higher in females than males, partly as a result of inhibition by androgens, stimulation by estrogen and depot-related differences in leptin expression. Leptin synthesis is greater in subcutaneous than in visceral adipose tissue, and the higher circulating concentration of leptin in females is likely to be due, in part, to a higher proportion of subcutaneous fat. Leptin has been implicated in other roles, including modulation of the reward circuitry for feeding, glucose metabolism, lipid oxidation, substrate partitioning, and adipocyte apoptosis [17, 19].
Adiponectin
The adiponectin gene on chromosome 3q27 was described in 1995. From the structural point of view, adiponectin is related to the complement 1q family and contains a carboxyl-terminal globular domain and an amino-terminal collagenous domain and also shares extensive sequence homology with collagen VIII and X [21]. Each monomer of adiponectin is composed of 3 domains: a variable N-terminal region, an α-helical collagenous ‘stalk’ composed of multiple G–X–X repeats, and a distinctive C-terminal globular domain of approximately 140 amino acids [19]. This adipokine circulates in three isoforms: a trimer, of low-molecular weight (LMW), a hexamer (trimer-dimer) of medium molecular weight (MMW) and a multimeric high molecular weight (HMW) isoform [22]. Adiponectin exists as a full-length protein (fAdp) of 30 kDa, which circulates in trimeric, hexameric and higher order complexes. A fragment containing the globular domain of adiponectin (gAdp) has also been shown to exhibit potent metabolic effects in various tissues. Adiponectin is secreted exclusively from adipose tissue [23]. There is a strong negative correlation between plasma adiponectin concentration in humans and fat mass, with the exception of severe cases of undernutrition and in the newborn [24]. Adiponectin is associated with type 2 diabetes (T2D), but is almost exclusively due to a decrease in levels of the circulating HMW isoform, without an accompanying reduction in levels of the other two oligomeric forms. The distribution of circulating adiponectin oligomers is thought to be primarily regulated at the stage of secretion from adipocytes, since interconversion between the different isoforms does not occur once they have been released from the cell [24]. In models of genetic and diet-induced obesity, adiponectin was shown to improve whole-body insulin sensitivity. Another role of adiponectin is to stimulate fatty acid oxidation and glucose uptake in skeletal muscle and adipose tissue; this effect is dependent on AMP-activated protein kinase (AMPK) signaling. Adiponectin is also involved in the suppression of hepatic glucose output through activation of AMPK [19].
Two receptors of adiponectin have been identified: AdipoR1 and AdipoR2. They contain 7 transmembrane domains, but differ structurally and functionally. Recent studies have shown that the skeletal muscle contains abundant levels of both AdipoR1 and AdipoR2 but the liver primarily expresses AdipoR2. AdipoR1 works with high affinity with gAdp and low affinity with fAdp, whereas AdipoR2 works with intermediate affinity with gAdp and fAdp. The biological effects of these receptors depend not only on blood concentrations of adiponectin but also tissue specificity [17, 25]. Adiponectin displays no great fluctuations in the bloodstream, which means that its release is not acute but regulated by long-term metabolic changes [25]. Adiponectin regulates energy expenditure through activation of AMPK in the hypothalamus, where AdipoR1 and AdipoR2 co-localize with the leptin receptor, ObR [17]. It has been demonstrated that adiponectin stimulated appetite and reduced energy expenditure and that these effects were eliminated following the ablation of AdipoR1 (siRNA) or AMPK signaling (AMPK dominant negative) [26].
In contrast to leptin, which has been suggested to enter the brain via endocytosis through the leptin receptor, the mechanism by which adiponectin is able to reach the hypothalamus is unknown. This study also showed an important finding: adipo–/– mice show markedly increased leptin sensitivity. This fact led to the proposal that the central actions of leptin and adiponectin have reciprocal functions for providing a homeostatic mechanism to maintain fat levels and energy stores through the suppression or stimulation of appetite and energy expenditure [19].
Tumor necrosis factor α
Tumor necrosis factor, TNF-α, is synthesized as a 26 kDa transmembrane protein that undergoes cleavage by a metalloproteinase to be released into the circulation as a 17 kDa soluble TNF-α molecule [10].
Adipocytes (isolated and differentiated) are capable of producing TNF-α. For some years it was suggested that adipocytes are the principal source of elevated TNF-α levels in obesity [21]. Nevertheless, more recently it has been recognized that adipocytes are not the major source of inflammatory cytokine but that macrophages from the stromal vascular fraction are the primary source of adipose derived TNF-α. Macrophages, which constitute about 10% of the stromal vascular fraction, are present in larger quantities in visceral adipose tissue than in subcutaneous adipose tissue [6]. These studies also postulate that the increased levels of this TNF-α in obesity are due to the increased infiltration of adipose tissue with M1 macrophages [21]. Some studies showed that the macrophages are formed as a result of transformation from monocytes that infiltrated the adipose tissue from the circulatory system [27]. It was also evidenced that, both in humans and in mice, the quantity of macrophages in the adipose tissue correlates with the fat mass. It was observed that infiltration of monocytes from the circulatory system is contributed by adipocytes, which by secreting monocyte chemotactic protein (MCP-1), macrophage migration inhibition factor (MIF-1), macrophage inflammatory proteins (MIP-1α), chemokine CCL5 (RANTES) and macrophage colony stimulating factor (M-CSF), support diversification and maturation of monocytes [27, 28].
The first adipose derived factor suggested to represent a link between obesity, inflammation and diabetes was TNF-α. Studies show that mRNA expression levels of TNF-α in adipose tissue in obesity is strongly implicated in the pathogenesis of insulin resistance; this is because it has been demonstrated that TNF-α can impair insulin signaling in hepatocytes and adipose tissue [29]. Other studies demonstrated that chronic treatment with TNF-α decreased insulin-stimulated glucose uptake in rat skeletal muscle, and targeted deletion of TNF-α or its receptors increased insulin sensitivity and glucose tolerance in obese rodents in some, but not all, studies ( Figure 4 ) [19].
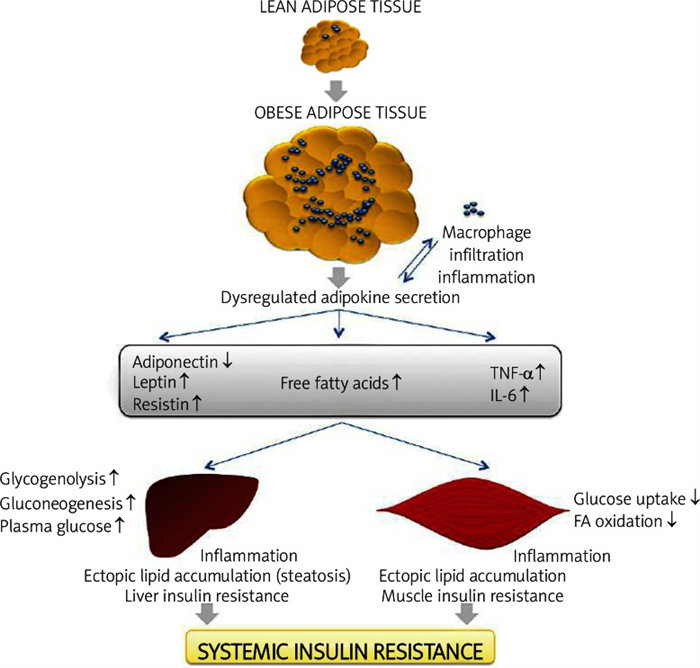
The expansion of adipose tissue leads to adipocyte hypertrophy in obesity. The release of che mokines that induce recruitment of macrophages from the bloodstream increases infiltration and inflammation with enhanced production of pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α) and IL-6. This is accomplished by increased release of FFA and dysregulated secretion of leptin, adiponectin and resistin. The macrophage and adipose tissue-derived adipokines acts in a paracrine or autocrine way, which exacerbates adipose tissue inflammation. Altered adipokine secretion, at the systemic level, can lead to decreased muscle and liver insulin sensitivity through enhanced ectopic lipid deposition and inflammation. These effects lead to increased liver glucose production (by means of gluconeogenesis and glycogenolysis). In contrast, muscle metabolism is reshuffled to a pattern of low glucose uptake and low FFA oxidation (with increases in levels of glycerol substrate for liver gluconeogenesis). These events lead to an increase of plasma glucose and, subsequently, an increase of insulin resistance. Adapted from: Galic S et al., 2010
TNF-α neutralization in obese T2D humans does not appear to improve glucose tolerance or insulin sensitivity. However, in individuals without established T2D, prolonged treatment does improve insulin sensitivity [19]. The molecular mechanism for this observed impairment in insulin action involves inhibition of insulin receptor substrate (IRS) signaling capability through the activation of serine kinases such as the c-Jun-N-terminal kinase (JNK) or inhibitor of NF-κB kinase (IKK) and through increased expression of suppressor of cytokine signaling 3 (SOCS3). In hepatocytes TNF-α also reduces fatty acid oxidation and skeletal muscle through effects mediated by the induction of protein phosphatase 2C and suppression of AMPK. The reduced rates of fatty acid oxidation are accompanied by increased accumulation of bioactive lipids, such as diacylglycerols, which in turn are known to activate protein kinase C and inhibit IRS function [19]. In addition, TNF-α induces pro-apoptotic and/or death signals in a variety of cell types. It is therefore interesting to speculate that hypertrophied adipocytes, which are stimulated by macrophage-derived TNF-α, can release saturated fatty acids as an endogen danger signal that report their diseased state to macrophages in obese adipose tissue. Indeed, several lines of evidence indicate that adipocyte death and/or the death receptor Fas signaling contribute to the obesity-induced adipose tissue, particularly during periods of starvation, but recent evidence has suggested pathophysiological roles other than the supply of nutrients in times of fasting or increased demand. In this regard free fatty acids, when released physiologically during fasting or starvation via adipocyte lipolysis, may not act as a danger signal [30].
Interleukin-6 (IL-6)
In humans, approximately 30% of circulating IL-6 originates from adipose tissue. Concentrations are higher in visceral fat as compared to subcutaneous fat. They increase with obesity and are stimulated by TNF and interleukin-1 (IL-1). Elevated levels are associated with increased risk of coronary artery disease, atherosclerosis, and unstable angina [31].
In T2D plasma IL-6 levels are increased and are positively correlated with body mass and plasma free fatty acid concentrations. As with TNF-α, the largest amount of IL-6 is derived from cells of the stromal vascular fractions, while the other part, approximately 1/3, of IL-6 detected in plasma is attributed to the production from white adipose tissue [2, 19]. It has been demonstrated that IL-6 inhibits the insulin signaling pathway by up-regulating SOCS3 expression, which in turn is known to impair insulin-induced insulin receptor and IRS-1 phosphorylation in adipocytes and hepatocytes. Moreover, IL-6 can promote fatty acid oxidation and glucose uptake in skeletal muscle findings, which are also observed with the IL-6 family member ciliary neurotrophic factor (CNTF) [32, 33]. Some studies have demonstrated that these effects require activation of AMPK-activated protein kinase but this mechanism is not understood [32]. In general, IL-6 inhibits lipase lipoprotein, induces lipolysis and increases glucose uptake [19].
Angiotensin
Adipose tissue expresses all components of the renin-angiotensin-aldosterone system (RAAS), including angiotensinogen (AGT), renin, angiotensin I-converting enzyme, and angiotensin II type 1 receptor [3]. Moreover, adipose tissue angiotensinogen mRNA and protein levels are regulated by nutrition, leading to decreased levels with fasting and to increased levels with refeeding. Angiotensin II stimulates prostacyclin synthesis, adipocyte differentiation and lipogenesis. Based on these findings, it is suggested that adipose tissue-derived angiotensin may regulate adipocyte differentiation and growth, as is the case in other tissues. It is also possible that RAAS peptides secreted by adipose tissue act on the vasculature and distant targets to regulate blood pressure and cardiovascular responses in obese individuals [32].
Angiotensin II also has a recognized effect on cardiovascular function such as hypertension and hemostasis. Human platelets express angiotensin II receptors AT1 [34]. Some studies have described the role of angiotensin II on hemostasis. It has been demonstrated that angiotensin II can induce and potentiate shape change in human platelets [35].
Plasminogen activator inhibitor 1 (PAI-1)
Another factor involved in microvascular events is the plasminogen activator inhibitor 1 (PAI-1).The gene for PAI-1 is located on chromosome 7q21.3- q22. PAI-1 is a single chain 45-kDa glycoprotein that contains from 379 to 381 amino acids. Endothelial and vascular smooth muscle cells are presumably the main sources of PAI-1 but other cells, such as platelets, hepatocytes, mesangial cells, fibroblasts, monocytes, macrophages, adipocytes, and stromal cells permeating the adipose tissue, have also been shown to secrete the serpin [36].
The greater the fat cell size and the adipose tissue mass, the greater is the contribution of adipose production to circulating PAI-1. Experimental data show that visceral adipose tissue has a higher capacity to produce PAI-1 than subcutaneous adipose tissue. Studies in human adipocytes indicate that PAI-1 synthesis is upregulated by insulin, glucocorticoids, angiotensin II, some fatty acids and, most potently, by cytokines such as TNF-α and transforming growth factor-β, whereas catecholamines reduce PAI-1 production [36, 37].
The PAI-1 is a protein involved in fibrinolysis and is altered in obesity [38]. Plasma PAI-1 levels increase in proportion to visceral adiposity, raising the possibility that PAI-1 serves as the link between abdominal/central obesity and cardiovascular disease. That protein can change the balance between fibrinolysis and fibrinogenesis, contributing to the remodeling of vascular architecture and the atherosclerotic process [39]. An altered function of the endocrine system and an impaired auto-/paracrine function at the fat cell levels may mediate this disturbance of the fibrinolytic system and thereby increase the risk for cardiovascular disease [37].
Acylation stimulating protein (ASP)
The ASP is produced through a two-step process involving three proteins of the alternate complement system: C3, factor B and adipsin, all of which are synthesized and secreted by adipocytes [40].
It has an important effect on the increase of lipogenesis by the translocation of glucose transporter type 4 (GLUT4) in glycerol 3-phosphate and the activity of diacylglycerol acyltransferase (DGAT), an enzyme catalyzing the synthesis of triglycerides [40, 41]. Plasma ASP increases with meals and facilitates the synthesis and storage of triglycerides. Consistent with its role as a mediator of lipogenesis, ASP deficiency increases postprandial fatty acid levels and decreases weight gain and triglyceride synthesis [39].
In humans, ASP levels are increased in obesity, T2D, and cardiovascular disease, whereas exercise or weight loss decreases ASP levels. Postprandially, subcutaneous adipose tissue increases production of ASP, and this has been shown to correlate with local fatty acid trapping. Furthermore, similar to insulin resistance, a deleterious ASP-resistant state has been proposed to also contribute to the disturbed adipose tissue metabolism and dyslipidemia common to diabetes and cardiovascular disease. The ASP levels curiously are also reduced with age, a phenomenon observed previously in humans, with children tending to have higher ASP values than adults [41, 42].
Resistin
Resistin, discovered in 2001, is a small peptide (12.5 kDa) synthesized as a peptide with 108 amino acids, containing high quantities of cysteine [19, 20]. The structure of resistin is strikingly similar to that of adiponectin [43]. Resistin is secreted not only by adipocytes, but also by a large number of cells, in particular immunocompetent cells.
Circulating resistin levels are increased in mouse models of obesity and in obese humans and are decreased by the anti-diabetic drug rosiglitazone, and increased in diet-induced and genetic forms of obesity, and administration of anti-resistin antibody has been shown to improve blood sugar and insulin action in mice with diet-induced obesity [44, 45]. Similarly, resistin has been implicated in the pathogenesis of diabetic complications and diabetes [45, 46].
In vitro studies evidenced that macrophage stimulation with lipopolysaccharide or pro-inflammatory cytokines (IL-1, IL-6 and TNF) leads to considerable increase in resistin production during infection [20].
Release of resistin appears to be stimulated by inflammation, LPS (lipopolysaccharide), IL-6, hyperglycemia, growth and gonadal hormones. While released within the fat tissue resistin acts on adipocytes themselves, leading to insulin resistance [45].
Effects of the administration and neutralization of resistin in glucose tolerance in tissues indicate that its action is through the negative modulation of one or more steps of insulin signaling aimed at increasing glucose uptake [44]. In addition, it promotes insulin resistance by increasing hepatic gluconeogenesis. It was also observed that the expression is about 3 times higher in pre-adipocytes compared to mature adipocytes, indicating that it is a potential regulator of adipogenesis [44, 47].
Resistin also stimulates endothelial cells to secrete such substances as monocyte chemoattractant protein-1 (MCP-1), vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1), which is indicated to be an adiponectin antagonist [27].
Visfatin
Visfatin is a highly conserved 52 kDa protein prevalent in visceral adipose tissue. It is also known as pre-B cell colony-enhancing factor (PBEF), is principally produced by adipocytes, although also by macrophages of the visceral adipose tissue, and in small quantities by subcutaneous adipose tissue. Visfatin mRNA expression significantly increases during differentiation of preadipocytes to adipocytes [48, 49].
It was shown that visfatin’s major function is related to energy metabolism and innate immunity and it is now regarded as a pro-inflammatory adipocytokine. Its properties induce activation of leukocytes and stimulate production of TNF-α and IL-6 [49, 50].
Inside cells visfatin acts as a nicotinamide phosphoribosyltransferase involved in the salvage pathway of NAD + biosynthesis. Thus, it is able to regulate cellular levels of NAD + , exerting an influence on cell energy metabolism and the activity of NAD + /NADH dependent enzymes [39].
Recent studies demonstrated that visfatin exerted insulin mimetic effects in cultured adipocytes, myocytes, and hepatocytes and lowered plasma glucose levels in mice by binding to and activating the insulin receptor [51].
Conclusions
Adipose tissue is the primary storage site for excess energy but it is also recognized as an endocrine organ. Adipocytes are now generally accepted to be a complex cell type involved in generating a number of signals which include cytokines, hormones and growth factors that not only affect the neighboring cells but also impact target tissues involved in energy metabolism and influencing physiologic and pathologic processes. Much of the research in this area has focused on leptin and adiponectin, the two prototypic adipokines, which show beneficial effects on insulin action and lipid metabolism. Obesity is characterized by increases in fat cell number, fat cell size, or a combination of the two. More recently, there is evidence that low-grade inflammation within the adipose tissue results in the dysregulation of adipocytokine production, thereby contributing to the pathophysiology of MetS. In the obese state, the adipose tissue is infiltrated by inflamed macrophages that release TNF-α and IL-6, thus linking obesity, inflammation and insulin resistance. It is increasingly important to understand the signaling pathways by which adipokines control metabolism and to try to discover new therapies for diseases related to adipose tissue.
References
1. Ottaviani E, Malagoli D, Franceschi C. The evolution of the adipose tissue: a neglected enigma. Gen Comp Endocrinol. 2011; 174 :1–4. [PubMed] [Google Scholar]
2. Bernlohr DA, Jenkins AE, Bennaars AA. Adipose tissue and lipid metabolism. In: Vence JE, Vence D, editors. Biochemistry of lipids, lipoproteins and membranes. 4th ed. Elsevier Science: Amsterdam; 2002. pp. 263–89. [Google Scholar]
3. Ahima RS, Flier JS. Adipose tissue as an endocrine organ. Trends Endocrinol Metab. 2000; 11 :327–32. [PubMed] [Google Scholar]
4. Fonseca-Alaniz MH, Takada J, Alonso-Vale MIC, Lima FB. Adipose tissue as an endocrine organ: from theory to practice. J Pediatr. 2007; 83 :S192–203. [PubMed] [Google Scholar]
5. Saely C, Geiger K, Drexel H. Browns versus white adipose tissue: a mini-review. Gerontol. 2012; 58 :120–2. [PubMed] [Google Scholar]
6. Trzeciak-Ryczek A, Tokarz-Deptuła B, Niedźwiedzka-Rystwej P, Deptula W. Adipose tissue – component of the immune system. Centr Eur J Immunol. 2011; 36 :95–9. [Google Scholar]
7. Costa JV, Duarte JS. Adipose tissue and adipokines [Portugues] Acta Med Port. 2006; 19 :251–6. [PubMed] [Google Scholar]
8. Fonseca-Alaniz MH, Takada J, Alonso-Vale MI, Lima FB. The adipose tissue as a regulatory center of the metabolism. Arq Bras Endocrinol Metabol. 2006; 50 :216–29. [PubMed] [Google Scholar]
9. Kiess W, Petzold S, Topfer M, et al. Adipocytes and adipose tissue. Best Pract Res Clin Endocrinol Metab. 2008; 22 :135–53. [PubMed] [Google Scholar]
10. Laclaustra M, Corella D, Ordovas JM. Metabolic syndrome pathophysiology: the role of adipose tissue. Nutr Metab Cardiovasc Dis. 2007; 17 :125–39. [PubMed] [Google Scholar]
11. Gray SL, Vidal-Puig AJ. Adipose tissue expandability in the maintenance of metabolic homeostasis. Nutr Rev. 2007; 65 :7–12. [PubMed] [Google Scholar]
12. Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO reports. 2001; 2 :282–6. [PMC free article] [PubMed] [Google Scholar]
13. Matsuzawa Y. The metabolic syndrome and adipocytokines. FEBS Letters. 2006; 580 :2917–21. [PubMed] [Google Scholar]
14. Yoneshiro T, Aita S, Matsushita M, Kameya T, Nakada K. Brown adipose tissue, whole-body energy expenditure, and thermogenesis in healthy adult men [Internet] Obesity. 2011; 19 :13–6. [PubMed] [Google Scholar]
15. Seneff S, Wainwright G, Mascitelli L. Is the metabolic syndrome caused by a high fructose, and relatively low fat, low cholesterol diet? Arch Med Sci. 2011; 7 :8–20. [PMC free article] [PubMed] [Google Scholar]
16. Stępień M, Wlazeł RN, Paradowski M, et al. Serum concentrations of adiponectin, leptin, resistin, ghrelin and insulin and their association with obesity indices in obese normo- and hypertensive patients – pilot study. Arch Med Sci. 2012; 8 :431–6. [PMC free article] [PubMed] [Google Scholar]
17. Itoh M, Suganami T, Hachiya R, Ogawa Y. Adipose tissue remodeling as homeostatic inflammation. Int J Inflam. 2011; 2011 :720926. [PMC free article] [PubMed] [Google Scholar]
18. Stepien M, Rosniak-Bak K, Paradowski M, et al. Waist circumference, ghrelin and selected adipose tissue-derived adipokines as predictores of insulin resistance in obese patients: preliminary results. Med Sci Monit. 2011; 17 :13–8. [PMC free article] [PubMed] [Google Scholar]
19. Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol. 2010; 316 :129–39. [PubMed] [Google Scholar]
20. Trzeciak-Ryczek A, Tokarz-Deptula B, Deptula W. Adipocytokines affecting the immune system – selected data. Centr Eur J Immunol. 2011; 36 :92–4. [Google Scholar]
21. Weisberg SP, Hunter D, Huber R, Lemieux J, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006; 116 :115–24. [PMC free article] [PubMed] [Google Scholar]
22. Kaser S, Tatarczyk T, Stadlmayr A, et al. Effect of obesity and insulin sensitivity on adiponectin isoform distribution. Eur J Clin Invest. 2008; 38 :827–34. [PubMed] [Google Scholar]
23. Palanivel R, Fang X, Park M, et al. Globular and full-length forms of adiponectin mediate specific changes in glucose and fatty acid uptake and metabolism in cardiomyocytes. Cardiovasc Res. 2007; 75 :148–57. [PubMed] [Google Scholar]
24. Schraw T, Wang ZV, Halberg N, Hawkins M, Scherer PE. Plasma adiponectin complexes have distinct biochemical characteristics. Endocrinology. 2008; 149 :2270–82. [PMC free article] [PubMed] [Google Scholar]
25. Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005; 26 :439–51. [PubMed] [Google Scholar]
26. Kubota N, Yano W, Kubota T, Yamauchi T. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007; 6 :55–68. [PubMed] [Google Scholar]
27. Ouchi N, Parker J, Lugus J, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011; 11 :85–97. [PMC free article] [PubMed] [Google Scholar]
28. Schäffler A, Schőlmerich J. Innate immunity and adipose tissue biology. Trends Immunol. 2010; 31 :228–35. [PubMed] [Google Scholar]
29. Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005; 11 :183–90. [PMC free article] [PubMed] [Google Scholar]
30. Suganami T, Ogawa Y. Adipose tissue macrophages: their role in adipose tissue remodeling. J Leukoc Biol. 2010; 88 :33–9. [PubMed] [Google Scholar]
31. Diamond F. The endocrine function of adipose tissue. Growth Genetics Horm. 2002; 18 :17–23. [Google Scholar]
32. Carey AL, Steinberg GR, Macaulay SL, et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes. 2006; 55 :2688–97. [PubMed] [Google Scholar]
33. Steinberg GR, Watt MJ, Ernst M, Birnbaum MJ, Kemp BE, Jørgensen SB. Ciliary neurotrophic factor stimulates muscle glucose uptake by a PI3-kinase-dependent pathway that is impaired with obesity. Diabetes. 2009; 58 :829–39. [PMC free article] [PubMed] [Google Scholar]
34. Touyz R, Schiffrin E. Effects of angiotensin II and endothelin-1 on platelet aggregation and cytosolic pH and free Ca 2+ concentrations in essential hypertension. Hypertension. 1993; 22 :853–62. [PubMed] [Google Scholar]
35. Jagroop IA, Mikhailidis DP. Angiotensin II can induce and potentiate shape change in human platelets: effect of losartan. J Hum Hypertens. 2000; 14 :581–5. [PubMed] [Google Scholar]
36. Correia MLG, Haynes WG. A Role for plasminogen activator inhibitor-1 in obesity: from Pie to PAI? Arterioscler Thromb Vasc Bio. 2006; 26 :2183–5. [PubMed] [Google Scholar]
37. Skurk T, Hauner H. Obesity and impaired fibrinolysis: role of adipose production of plasminogen activator inhibitor-1. Int J Obese Relat Metab Disord. 2004; 281 :357–64. [PubMed] [Google Scholar]
38. Mertens I, Van Gaal L. Visceral fat as a determinant of fibrinolysis and hemostasis. Semin Vasc Med. 2005; 5 :48–55. [PubMed] [Google Scholar]
39. Manolescu B, Stoian I, Atanasiu V, Busu C, Lupescu O. The role of adipose tissue in uraemia-related insulin resistance. Nephrology (Carlton) 2008; 13 :622–8. [PubMed] [Google Scholar]
40. Cianflone K, Xia Z, Ying L. Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim Biophys Acta. 2003; 1609 :127–43. [PubMed] [Google Scholar]
41. Paglialunga S, Fisette A, Yan Y, et al. Acylation-stimulating protein deficiency and altered adipose tissue in alternative complement pathway knockout mice. Am J Physiol Endocrinol Metab. 2008; 29 :E521–9. [PubMed] [Google Scholar]
42. Cianflone K, Lu H, Smith J, Yu W, Wang H. Adiponectin, acylation stimulating protein and complement C3 are altered in obesity in very young children. Clin Endocrinol. 2005; 62 :567–72. [PubMed] [Google Scholar]
43. Patel SD, Rajala MW, Rossetti L, Scherer PE, Shapiro L. Disulfide-dependent multimeric assembly of resistin family hormones. Science. 2004; 304 :1154–8. [PubMed] [Google Scholar]
44. Steppan C, Bailey S, Bhat S, Al E. The hormone resistin links obesity to diabetes. Nature. 2007; 409 :307–12. [PubMed] [Google Scholar]
45. Guzik TJ, Mangalat D, Korbut R. Adipocytokines – novel link between inflammation and vascular function? J Physiol Pharmacol. 2006; 57 :505–28. [PubMed] [Google Scholar]
46. Wasim H, Al-Daghri N, Chetty R, McTernan P, Barnett A, Kumar S. Relationship of serum adiponectin and resistin to glucose intolerance and fat topography in South-Asians. Cardiovasc Diabetol. 2006; 5 :10. [PMC free article] [PubMed] [Google Scholar]
47. Mcternan P, Mcternan C, Chetty R, et al. Increased resistin gene and protein expression in human abdominal adipose tissue. J Clin Endocrinol Metab. 2002; 87 :2407–10. [PubMed] [Google Scholar]
48. Olszanecka-Glinianowicz M, Kocelak P, Nylec M, Chudek J, Zahorska-Markiewicz B. Circulating visfatin level and visfatin/insulin ratio in obese women with metabolic syndrome. Arch Med Sci. 2012; 8 :214–21. [PMC free article] [PubMed] [Google Scholar]
49. Varma V, Yao Borengasser A, Rasouli N, Al E. Human vistatin expression: relationship to insulin sensitivity, intramyocellular lipids, and inflammation. J Clin Endocrinol Metab. 2007; 92 :666–72. [PMC free article] [PubMed] [Google Scholar]
50. Moschen A, Kaser A, Enrich B, Al E. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007; 178 :1748–58. [PubMed] [Google Scholar]
51. Wang P, Xu TY, Guan YF, Su DF, Fan GR, Miao CY. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovas Res. 2009; 81 :370–80. [PubMed] [Google Scholar]